
¿Qué es un «nuevo dispositivo» según el MDR y el IVDR?
🩺 Introducción
El Reglamento de Productos Sanitarios (MDR, UE 2017/745) y el Reglamento sobre Diagnósticos In Vitro (IVDR, UE 2017/746) han endurecido significativamente los requisitos para los productos sanitarios y los diagnósticos in vitro.
En este contexto, surge la pregunta: ¿Qué se considera un «nuevo dispositivo» bajo el MDR y el IVDR? ¿Qué criterios y requisitos deben tener en cuenta los fabricantes?
🧾 Definición de «nuevo dispositivo» según el MDR y el IVDR
Los términos «nuevo dispositivo» o «nuevo producto sanitario» no están definidos explícitamente en el MDR ni en el IVDR.
Sin embargo, un producto sanitario se considera nuevo si:
✅ Utiliza un diseño o tecnología completamente nueva
✅ Tiene una nueva finalidad médica o nuevas indicaciones
✅ Representa una modificación fundamental de un producto existente
✅ Se introduce por primera vez en el mercado europeo
✅ No ha sido previamente clasificado bajo el MDR o el IVDR
🚫 No se considera un nuevo dispositivo:
❌ Un producto con modificaciones menores de un diseño existente
❌ Una simple actualización o mejora de un dispositivo ya aprobado
❌ Un producto que ya existe en una versión comparable en el mercado de la UE
🧪 Ejemplos de «nuevo dispositivo»
Productos sanitarios según el MDR:
- Nuevos implantes con combinaciones innovadoras de materiales
- Tecnología médica portátil con monitoreo de salud basado en IA
- Prótesis personalizadas impresas en 3D
- Sistemas quirúrgicos robotizados con nuevas funcionalidades
Diagnósticos in vitro según el IVDR:
- Pruebas moleculares con nuevas tecnologías de análisis
- Análisis de sangre para enfermedades previamente no diagnosticables
- Software de diagnóstico basado en IA para medicina personalizada
- Pruebas de biomarcadores innovadores con mayor sensibilidad y especificidad
📋 Requisitos reglamentarios para un «nuevo dispositivo»
Un nuevo dispositivo médico está sujeto a procedimientos estrictos de evaluación y aprobación, especialmente si no encaja en una clasificación de producto ya existente.
1. Clasificación del nuevo dispositivo
Los productos sanitarios se clasifican según el MDR en las clases I, IIa, IIb o III.
Para un nuevo dispositivo se realiza una evaluación de riesgos precisa: cuanto mayor es el riesgo, más estrictos son los requisitos normativos.
| Clase | Nivel de riesgo | Ejemplo |
|---|---|---|
| I | Bajo | Termómetros digitales |
| IIa | Medio | Instrumentos electroquirúrgicos |
| IIb | Alto | Respiradores |
| III | Muy alto | Marcapasos implantables |
2. Evaluación clínica y estudios de rendimiento
Se requieren datos clínicos sólidos para nuevos dispositivos, incluyendo:
- Estudios clínicos sobre seguridad y eficacia
- Datos comparativos con productos existentes
- Estudios de rendimiento para diagnósticos in vitro bajo el IVDR
3. Documentación técnica y marcado CE
Los fabricantes deben elaborar una documentación técnica completa conforme a los Anexos II y III del MDR/IVDR, que incluya:
- Descripción y especificaciones del producto
- Archivo de gestión de riesgos
- Evidencias de biocompatibilidad y seguridad
- Instrucciones de uso y etiquetado
4. Evaluación de la conformidad y organismos notificados
Según la clase del nuevo dispositivo, puede ser necesario contar con un organismo notificado para supervisar el proceso:
- Clase I: Autocertificación por parte del fabricante
- Clases IIa, IIb, III: El organismo notificado revisa la documentación técnica, los datos clínicos y los procesos de fabricación
⚠️ Retos para los fabricantes de nuevos dispositivos
📌 Plazos de aprobación más largos debido a los requisitos normativos reforzados
📌 Requisitos más estrictos de evidencia para demostrar seguridad y eficacia
📌 Costes elevados por estudios clínicos, evaluación de conformidad y vigilancia post-comercialización (PMS)
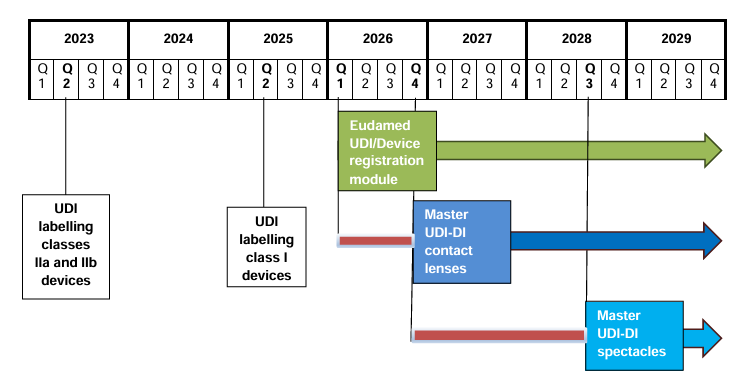
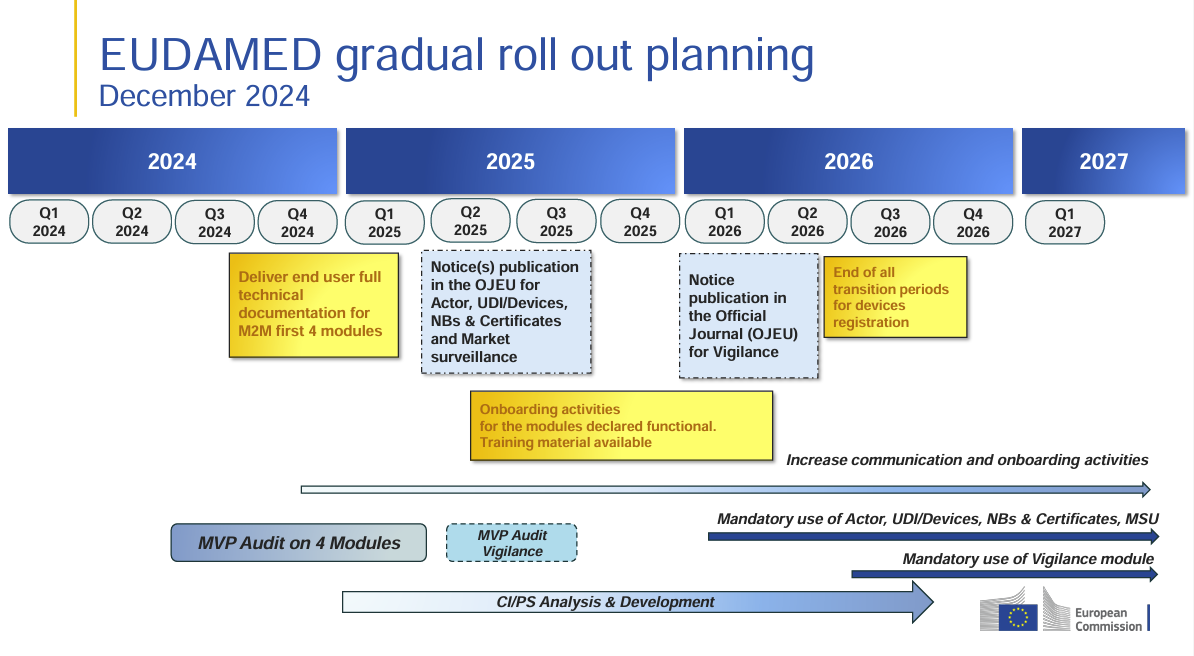
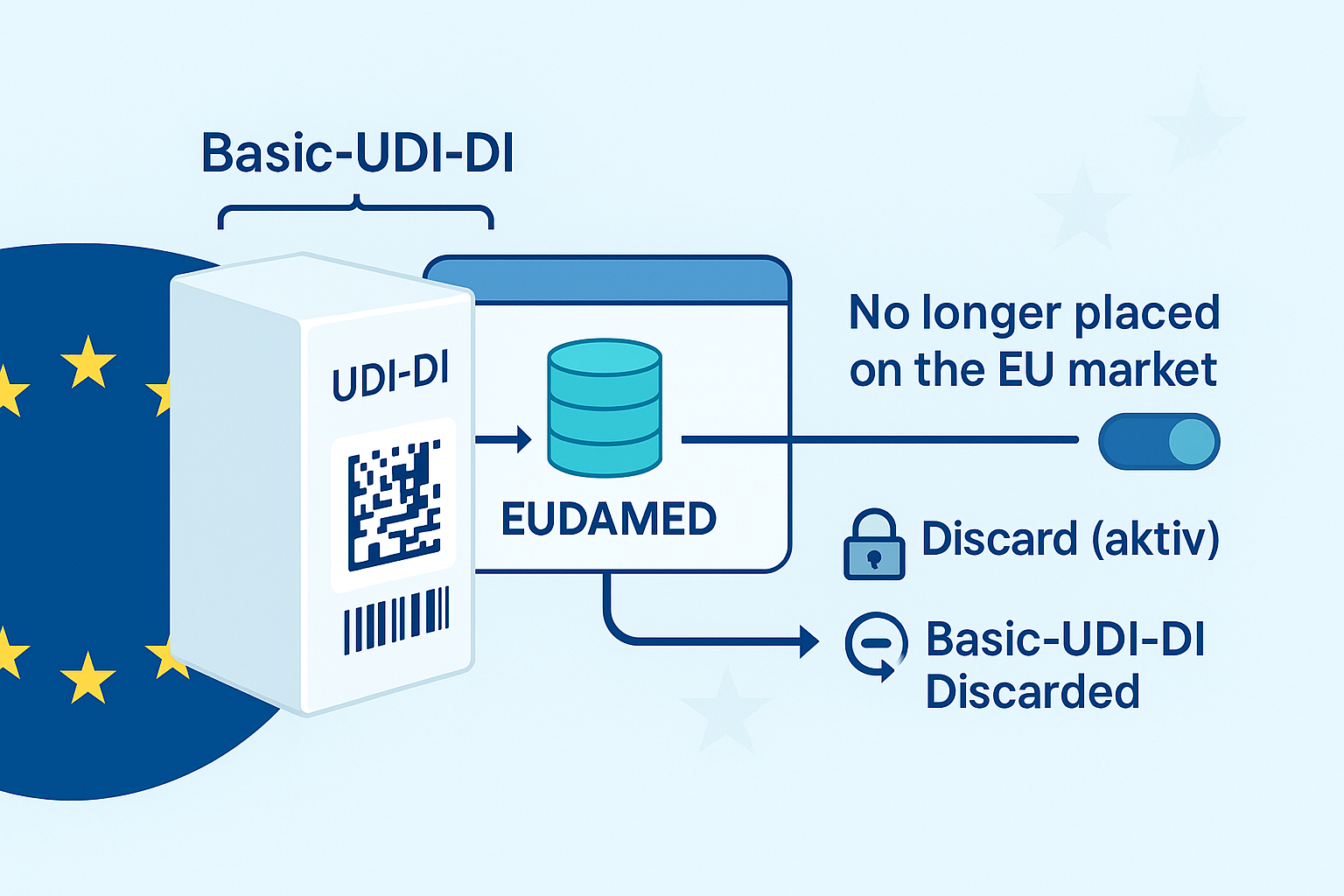
📌 Mayor control del mercado a través de EUDAMED y del sistema UDI
✅ Conclusión
Un «nuevo dispositivo» es un producto sanitario o diagnóstico in vitro que introduce tecnologías, indicaciones o materiales novedosos y, por tanto, no se corresponde con un dispositivo ya existente.
Estos productos están sujetos a una evaluación normativa rigurosa, especialmente en lo que respecta a la evaluación de riesgos, evidencia clínica y documentación técnica.
Los fabricantes deben integrar los requisitos del MDR y del IVDR desde las primeras etapas del desarrollo para lograr una aprobación eficiente del mercado.
📌 Fuentes:
- Reglamento (UE) 2017/745 sobre productos sanitarios (MDR)
- Reglamento (UE) 2017/746 sobre productos sanitarios para diagnóstico in vitro (IVDR)
- Documentos guía MDCG sobre clasificación y evaluación clínica
- Base de datos EUDAMED de productos sanitarios






Related Posts