
Términos UDI y atajos en la industria médica de la A a la Z que debes conocer.
A B C D E F G H I J K L M N O P Q R S T U V W X Y Z
A
ACKs (Acuses de recibo)
En el contexto de eMDR, hay tres acuses de recibo: Ack1 confirma la recepción de tu envío en el ESG (Electronic Submissions Gateway); Ack2 indica que el ESG descomprimió correctamente el archivo y lo remitió al CDRH; y Ack3 proporciona el resultado del procesamiento (aceptado/error). El ESG es el portal central de presentaciones de la FDA (actualmente ESG NextGen), y el CDRH es el Centro de Dispositivos y Salud Radiológica de la FDA.
Adverse Event Reporting (Notificación de eventos adversos)
Es el proceso exigido por la normativa mediante el cual los fabricantes y operadores registran, evalúan y notifican los eventos adversos (p. ej., efectos secundarios, incidentes graves) dentro de los plazos establecidos, para que las autoridades puedan detectar señales de seguridad e iniciar medidas como retiradas del mercado.
AIDC: Identificación Automática y Captura de Datos
Los sistemas AIDC pueden apoyar la producción, logística, transporte y distribución de bienes, hacerse cargo del seguimiento de bienes y dispositivos y verificar su marcado e identificación.
Las tecnologías AIDC incluyen el marcado gráfico de mercancías con códigos de barras y códigos 2D.
AIMDD
Directiva de Dispositivo Médico Implentable Activo (será reemplazada por MDR).
Protocolo AS2: Declaración de aplicabilidad 2
El protocolo AS2 es uno de los estándares de transmisión EDI (intercambio electrónico de datos) más populares. La razón de su uso generalizado en los negocios es que AS2 utiliza Internet como canal de comunicación. Por lo tanto, el protocolo no depende del uso de una red propietaria. Para la transmisión a través de Internet, AS2 utiliza protocolos HTTP y HTTPS.
Protocolo AS4: Declaración de aplicabilidad 4
El protocolo AS4 es un protocolo de mensajería basado en servicios web para el intercambio seguro de mensajes de empresa a empresa entre socios comerciales. El protocolo fue aprobado por el comité técnico de OASIS (Organización para el Avance de los Estándares de Información Estructurada) a ebXML Messaging Services. Con su capacidad de servicios web, AS4 tiene la oportunidad de convertirse en el estándar de comunicación basado en la nube.
AusUDID
La Base de Datos Australiana de Identificación Única de Dispositivos (AusUDID) es la base pública de UDI de la Therapeutic Goods Administration (TGA) de Australia.
Representante autorizado
Un representante autorizado es un representante que actúa y decide en nombre de otra persona mediante una declaración de intenciones o recibiéndola.
La definición, los derechos y los deberes de un representante autorizado se describen en el capítulo 11 del MDR.
Leer más sobre >> Representante autorizado de MDR <<B
Código UDI-DI básico
El UDI-DI BÁSICO resume las características comunes de un grupo de productos.
Budi
Se utiliza para acortar el UDI-DI BÁSICO.
C
CA: Autoridad Competente
Organismo responsable de la aprobación de dispositivos médicos.
Leer másCHRN
Número de registro único emitido por Swissmedic para los operadores económicos.
Investigación clínica
Se define como «cualquier investigación o estudio sistemático en o sobre uno o más sujetos humanos, realizado para evaluar la seguridad y/o el rendimiento de un dispositivo médico». (SG5/N1:2007).
Evalúa la seguridad y el rendimiento clínico del dispositivo en cuestión y evalúa si el dispositivo es adecuado para el (los) propósito(s) y la (s) población (es) para la (s) que está (n) destinado (s) (EN ISO 14155-1:2009).
Lea el capítulo VI del MDR para obtener más información sobre la investigación clínica relacionada con los dispositivos colocados en la UE.
CND: Classificazione Nazionale dei Dispositivi medici
El sistema italiano, en inglés «National Classification of Medical Devices», ha solicitado el registro de dispositivos médicos en EUDAMED. Allí se llama EMDN.
Código 128
El Code128 es un código de barras alfanumérico con alta densidad de información. La estructura principal consiste en un carácter de inicio, la información del usuario, el dígito de control y el carácter de parada. Un área blanca (campo brillante o zona tranquila) debe mantenerse libre antes del carácter de inicio y después del carácter de parada.
Advertencias críticas
Advertencias o precauciones que deben tomarse y que deben ponerse en conocimiento inmediato del usuario del dispositivo y de cualquier otra persona. Esta información podrá mantenerse al mínimo, en cuyo caso deberá aparecer información más detallada en las instrucciones de uso, teniendo en cuenta los usuarios previstos. (Fuente: Reglamento de dispositivos médicos 2017/745, Anexo I, 23.2 sobre la información que debe contener la etiqueta)
Por ejemplo: » si se ingiere: obtenga asesoramiento/atención médica inmediata», «explosivo cuando esté seco», «tóxico por contacto visual».
Para obtener más información, lea nuestro artículo Detalles sobre las condiciones de almacenamiento/manipulación y advertencias críticas
CSDT: Plantilla de expediente de envío común
Documento de la ASEAN destinado a proporcionar una plantilla común para la presentación de información sobre dispositivos médicos a las autoridades reguladoras de dispositivos médicos de los países miembros de la ASEAN. Aquí está la guía de la ASEAN sobre esta plantilla.
Dispositivo hecho a medida
– dispositivo fabricado específicamente de acuerdo con una prescripción escrita de cualquier persona autorizada por la legislación nacional en virtud de las cualificaciones profesionales de esa persona
– dispositivo que proporciona características de diseño específicas proporcionadas bajo la responsabilidad de esa persona
– producto destinado al uso exclusivo de un paciente en particular exclusivamente para satisfacer sus condiciones y necesidades individuales.
– Siempre que el producto no se «produzca en masa» (fabricado en grandes cantidades mediante un proceso mecánico automatizado), los fabricantes pueden utilizar tecnologías modernas de vanguardia para fabricar CMD.
Para obtener más información, lea nuestro artículo dedicado a este tema: Dispositivo hecho a medida (CMD) según MDR (2017/745).
D
Código de barras DataMatrix
El código DataMatrix es uno de los códigos 2D más conocidos. Se utiliza para el marcado directo permanente, pero también cada vez más como imagen de código impreso en el manejo de documentos.
Dimdi
El Instituto Alemán de Documentación e Información Médica era una autoridad subordinada del Ministerio Federal de Salud.
Distribuidor (según MDR)
La definición, los derechos y las obligaciones de los distribuidores se describen en el capítulo 14 del MDR.
Leer másDI: Identificador del dispositivo
Datos de identificación del producto (estáticos) para un dispositivo como GTIN.
DM: Marcado directo
Un número de identificación del dispositivo. Por ejemplo, número GTIN14 o número HIBC.
DPM: marcado directo de piezas
Marcado de un producto sin etiqueta. Por ejemplo, el grabado láser de un número de serie en instrumentos quirúrgicos.
DUNS: Sistema de Numeración Universal de Datos
El número DUNS es un código numérico desarrollado por Dun&Bradstreet y utilizado internacionalmente como estándar para la identificación única de empresas.
E
EMDN
La Nomenclatura Europea de Productos Sanitarios se ha consolidado como el sistema de registro en EUDAMED.
eMDR
Se refiere a la presentación electrónica de Informes de Dispositivos Médicos (MDR, por sus siglas en inglés) a la FDA de los Estados Unidos por parte de fabricantes, importadores y establecimientos sanitarios usuarios (user facilities).
ESG: Pasarela de Presentaciones Electrónicas
El Portal de Presentaciones Electrónicas de la Administración de Alimentos y Medicamentos es una solución para toda la agencia para la aceptación de presentaciones electrónicas. El ESG de la FDA es un punto de intercambio altamente escalable, fácilmente disponible, potente y seguro para que la FDA y sus socios manejen una variedad de documentos y presentaciones a través de protocolos estándar de la industria. El ESG de la FDA permite la presentación segura de información regulatoria previa y posterior a la comercialización para su revisión.
El ESG de la FDA ofrece dos métodos, WebTrader y AS2, para presentar solicitudes a la FDA.
EUDAMED: Base dedatos europea de dispositivos médicos
Base de datos para el registro de productos sanitarios en la Unión Europea. Una nueva versión de la base de datos estará disponible como parte de la introducción del nuevo Reglamento de Dispositivos Médicos.
EURL: Leboratorio de refracción de la UE
Los laboratorios designados por la Comisión Europea para emitir opiniones y puntos de vista sobre algunos dispositivos médicos de alto riesgo y div y tienen un papel consultivo para actores como la Comisión Europea o el MDCG. Las tareas del EURLS se definen en el artículo 100, apartados 2 y 3, del Reglamento sobre productos sanitarios para diagnóstico in vitro.
F
FDA
La FDA de los Estados Unidos es la administración de alimentos y medicamentos de los Estados Unidos. Como tal, informa al Departamento de Salud y Servicios Humanos de EE. UU.
Enlace: https://www.fda.gov/
FSCA: Acción Correctiva de Seguridad en Campo
Cualquier acción tomada por el propietario de un dispositivo para reducir el riesgo de muerte o deterioro grave de la salud asociado con el uso de un dispositivo médico que ya está en el mercado.
Con respecto al FSCA en el EUDAMED, consulte el artículo 87 del MDR y el artículo 82 del IVDR.
FSN (Aviso de Seguridad en el Campo)
Documento complementario de una FSCA: la notificación enviada a clientes/usuarios. Describe el problema, los productos afectados y ofrece instrucciones (p. ej., dejar de usar el producto).
G
RGPD
El Reglamento General de Protección de Datos es un reglamento de protección de datos introducido en la Unión Europea (UE). Entró en vigor el 25 de mayo de 2018, con el objetivo de reforzar y armonizar la protección de los datos personales de los ciudadanos de la UE. El RGPD establece normas estrictas sobre cómo se pueden recopilar, procesar, almacenar y transferir los datos personales.
Enlace: https://gdpr-info.eu/
GDSN
La Red Global de Sincronización de Datos (GDSN) es un sistema basado en Internet para la transmisión de datos utilizado en todo el mundo. GDSN garantiza que los datos intercambiados entre socios comerciales cumplan con los estándares GS1 en todo el mundo.
Enlace: https://www.gs1.org/services/gdsn
GHTF: Grupo de Trabajo de Armonización Global sobre Dispositivos Médicos
El Grupo de Trabajo de Armonización Global era «un grupo voluntario de representantes de las autoridades reguladoras nacionales de dispositivos médicos» cuyo objetivo era la estandarización mundial de la regulación de dispositivos médicos. Estaba formado por miembros de la industria.
El GHTF se disolvió a finales de 2012. Su papel fue asumido por el Foro Internacional de Reguladores de Dispositivos Médicos (IMDRF), una organización de seguimiento compuesta por funcionarios de las autoridades reguladoras, no de la industria, de todo el mundo. El sitio web de GHTF ya no funciona.
GMDN: Nomenclatura Global de Dispositivos Médicos
El código GMDN es necesario para registrar dispositivos médicos en la base de datos de la FDA llamada GUDID.
Enlace: https://www.gmdnagency.org/
GS1
GS1 es una red de organizaciones sin fines de lucro que desarrollan, negocian y mantienen estándares para procesos entre empresas en todo el mundo. Han sido designados como el organismo de asignación para la operación de un sistema para la asignación de UDI en el contexto de la identificación única de productos de dispositivos médicos.
Enlace: https://www.gs1.org/
GTIN
El Global Trade Item Number es un número de identificación derivado del sistema GS1 (anteriormente sistema EAN) con el que se pueden marcar las unidades de trading.
GUDID
La Base de Datos Global de Identificación Única de Dispositivos es una base de datos administrada por la FDA que sirve como catálogo de referencia para cada dispositivo con un identificador único de dispositivo (UDI).
Enlace a GUDID Productiv: https://gudid.fda.gov/gudid/app/login.xhtml
Enlace a GUDID PreProd: https://gudid.preprod.fda.gov/gudid/
H
HIBC: Código de barras de la industria de la salud
Número único mundial que sirve para la identificación de productos.
Enlace: https://www.hibcc.org/
HIPAA
Enlace: https://www.cdc.gov/phlp/publications/topic/hipaa.html
HL7
Health Level 7 es un grupo de normas internacionales para el intercambio de datos entre las organizaciones sanitarias y sus sistemas informáticos.
Leer másI
ICCBBA: Consejo Internacional para lo Común en la Automatización de los Bancos de Sangre
El ICCBBA es una organización de normalización internacional, sin ánimo de lucro y no gubernamental que tiene una relación oficial con la Organización Mundial de la Salud (OMS) y es responsable de la gestión y el desarrollo de la norma ISBT 128. Gestiona, desarrolla y licencia el estándar internacional de información para la terminología, codificación y etiquetado de dispositivos médicos de origen humano. El ICCBBA actúa como una agencia emisora en el campo del UDI.
Enlace: https://www.iccbba.org/
ICSR Report
Un ICSR (Individual Case Safety Report, Informe Individual de Seguridad) es un informe de seguridad estandarizado sobre un caso único de un evento adverso o un problema de producto. En la farmacovigilancia y la vigilancia de dispositivos médicos (p. ej., según ICH E2B(R3)), se intercambia electrónicamente para garantizar que los casos se notifiquen y evalúen de manera coherente.
IFA – Informationsstelle für Arzneispezialitäten
“Centro de información de especialidades farmacéuticas”
IFA GmbH fue designado como el organismo de asignación para UDI-DI por la Comisión de la UE en su decisión de implementación de 6 de junio de 2019. Un número de producto de farmacia (PPN) codificado en el paquete de acuerdo con el sistema de codificación IFA representa, por lo tanto, un UDI-DI compatible con MDR.
Enlace: http://www.ifaffm.de/en/home.html
IMDRF: Foro Internacional de Reguladores de Dispositivos Médicos
El Foro Internacional de Reguladores de Dispositivos Médicos se concibió en febrero de 2011 como un foro para identificar futuras directrices para la armonización de las regulaciones de dispositivos médicos.
Es un grupo voluntario de reguladores de dispositivos médicos de todo el mundo que se han unido para construir sobre la sólida base del Grupo de Trabajo de Armonización Global sobre Dispositivos Médicos (GHTF) y para acelerar la armonización y convergencia internacional de la regulación de dispositivos médicos.
Enlace: http://imdrf.org/
Importadores
La definición, los derechos y las obligaciones de los importadores se describen en el capítulo 13 del MDR.
Leer másOrganismo emisor
El Organismo Emisor es responsable de la operación de un sistema para la asignación de UDI en el marco de la identificación única de dispositivos para dispositivos médicos. Las agencias emisoras conocidas son GS1, HIBCC y otras.
Leer másIVD: Diagnóstico in vitro
Dispositivo médico de diagnóstico in vitro es un término para los dispositivos médicos utilizados para el examen de laboratorio médico de muestras tomadas del cuerpo humano. Estos se examinan fuera del cuerpo (latín in vitro ‘en vidrio’).
IvDD
Directiva de Diagnóstico In Vitro (a sustituir por IVDR).
IVDR: Regulación de diagnóstico in vitro
El nuevo Reglamento Europeo sobre dispositivos médicos de diagnóstico in vitro sustituye a la Directiva de Diagnóstico In Vitro existente.
Es válido desde el 25.05.2017.
J
JSON: Notación de objetos JavaScript
La notación de objetos de JavaScript es un formato de datos compacto en forma de texto fácil de leer y sirve para el intercambio de datos entre aplicaciones. JSON es independiente del lenguaje de programación. Existen analizadores y generadores en todos los idiomas comunes.
K
Kit (UE)
El kit se describe en el marco legal de la Unión Europea solo en el IVDR.
‘kit’ significa un conjunto de componentes que se empaquetan juntos y están destinados a ser utilizados para realizar un examen de diagnóstico in vitro específico, o una parte del mismo
Quelle: REGLAMENTO (UE) 2017/746, Capítulo 1, Artículo 2, Definiciones (Párrafo 11)
Kit (EE. UU.)
En la definición de la FDA, el kit o kit de conveniencia debe interpretarse como compuesto por dos o más dispositivos médicos diferentes empaquetados juntos para la comodidad del usuario.
La definición original se puede encontrar en el siguiente enlace
L
Látex
El material látex está contenido en muchos productos y siempre debe ser declarado. Puede causar una fuerte reacción alérgica en algunas personas y dañar su salud. Esta es una de las razones por las que este atributo siempre debe declararse al registrar productos médicos.
Dispositivo heredado
Dispositivos médicos, dispositivos médicos implantables activos y dispositivos médicos de diagnóstico in vitro que estén cubiertos por un certificado válido emitido de acuerdo con la Directiva 93/42/CEE, la Directiva 90/385/CEE o la Directiva 98/79/CE y que continúen comercializándose después de la fecha de aplicación del Reglamento (UE) 2017/745 (MDR) o el Reglamento 2017/746 (IVDR). Guía de la UE aquí.
Legislación
Las legislaciones europeas que están en el foco de la UDI son:
Reglamento de dispositivos médicos (MDR), Reglamento de diagnóstico in vitro (IVDR), Directiva de dispositivos médicos (MDD), Directiva de dispositivos médicos implantables activos (AIMDD), Directiva de diagnóstico in vitro (IVDD)
M
MASTER-UDI
La Comisión Europea está modificando el Reglamento (UE) 2017/745 para mejorar el sistema de identificación única de dispositivos (UDI) para lentes de contacto y, en una etapa posterior, para productos que pueden tener muchas variaciones.El sistema actual requiere que los fabricantes asignen un UDI a cada variante de lentes de contacto, lo que ha llevado a un número abrumador de UDI-DI (Identificadores de Dispositivos) en la Base de Datos Europea de Dispositivos Médicos (Eudamed).Para abordar este problema, la Comisión ha desarrollado el concepto «Master UDI-DI«, que agrupa las lentes de contacto con las mismas combinaciones de parámetros clínicos y de diseño en un solo identificador. Esta modificación hará que el proceso de identificación sea más eficiente, sin comprometer la seguridad o la trazabilidad.
La nueva regulación se aplicará dos años después de su entrada en vigor, pero los fabricantes pueden comenzar a usar el UDI-DI Maestro y el UDI-PI (Identificador de Producción) antes si lo desean.Puedes encontrar más información aquí: EUDAMED MASTER-UDI
Fabricante (según MDR)
La definición, los derechos y las obligaciones de los fabricantes se explican en el capítulo 10 del MDR.
Leer más
MDCG: Grupo de Coordinación de Dispositivos Médicos
El Grupo de Coordinación de Productos Sanitarios asesora a la Comisión Europea y apoya a la Comisión y a los Estados miembros para garantizar una aplicación armonizada de los Reglamentos (UE) 2017/745 y 2017/746 sobre productos sanitarios.
MDD
Directiva de Dispositivos Médicos (será reemplazada por MDR).
MDR: Regulación de Dispositivos Médicos
El nuevo Reglamento Europeo de Productos Sanitarios sustituye a las directivas existentes sobre productos sanitarios.
Es válido desde el 25.05.2017.
MDSAP: Programa de Auditoría Única de Dispositivos Médicos
Permite que una Organización de Auditoría reconocida por MDSAP realice una sola auditoría regulatoria de un fabricante de dispositivos médicos que satisfaga los requisitos relevantes de las autoridades regulatorias que participan en el programa.
Los miembros son: Australia, Brasil, Canadá, Japón, FDA. la UE, Reino Unido, y QUE son solo observadores oficiales.
MIR (Manufacturer Incident Report)
Notificación de un incidente grave (muerte, deterioro grave de la salud o riesgo para la salud pública).
M2M: Machine-to-Machine
Machine-to-machine significa el intercambio automatizado de información entre dispositivos terminales como máquinas, máquinas expendedoras, vehículos o contenedores entre sí o con un centro de control central, utilizando cada vez más Internet y las diversas redes de acceso, como la red de telefonía móvil.
MTR (Informe de Tendencias del Fabricante)
Notificación a la autoridad cuando el fabricante detecta un aumento significativo (tendencia) en incidentes no graves o efectos secundarios esperados.
N
NANDO
Organizaciones Notificadas y Designadas de Nuevo Enfoque (Organismo Notificado, por ejemplo TÜV Süd).
Enlace: https://ec.europa.eu/growth/tools-databases/nando/
NDC: Código Nacional de Medicamentos
El Código Nacional de Medicamentos (NDC) es un identificador de producto único utilizado en los Estados Unidos para medicamentos destinados al uso humano. La Ley de Listado de Medicamentos de 1972 requiere que los establecimientos de medicamentos registrados proporcionen a la Administración de Alimentos y Medicamentos (FDA) una lista actualizada de todos los medicamentos fabricados, preparados, propagados, compuestos o procesados por ella para su distribución comercial. Los productos farmacéuticos se identifican e informan utilizando el NDC.
Organismo notificado
Los Organismos Notificados de la Unión Europea son organismos privados de inspección (organismos de auditoría y certificación) notificados y supervisados por el Estado, que actúan en nombre de los fabricantes con el fin de acompañar y controlar la evaluación de la conformidad de los fabricantes de productos industriales de diversa índole.
Leer másO
Reconocimiento óptico de caracteres (OCR)
El OCR es una tecnología utilizada para convertir diferentes tipos de documentos, como documentos en papel escaneados, archivos PDF o imágenes capturadas por una cámara digital, en datos editables y de búsqueda. En el contexto de la UDI y el etiquetado, la OCR podría utilizarse para capturar y digitalizar información de las etiquetas.
Propietario/Operador
En las regulaciones de UDI de la FDA de EE. UU., este término se refiere a la entidad responsable de fabricar, reempaquetar, reetiquetar y distribuir un dispositivo.
Uso no indicado en la etiqueta
Esto se refiere al uso de un dispositivo médico (o fármaco) para una indicación no aprobada o en un grupo de edad, dosis o vía de administración no aprobados. Este concepto puede surgir en las discusiones sobre el etiquetado, ya que las etiquetas deben reflejar con precisión los usos aprobados de un producto.
OEM (Fabricante de equipos originales)
En el contexto de los dispositivos médicos, un OEM es una empresa que produce piezas y equipos que pueden ser comercializados por otro fabricante. El OEM también puede ser responsable de ciertos aspectos de UDI y etiquetado, dependiendo de los requisitos reglamentarios.
Gestión de la obsolescencia
Esto se refiere a la práctica de garantizar que un dispositivo cumpla con las regulaciones actuales a lo largo de su ciclo de vida, incluso cuando algunos componentes o materiales utilizados en su construcción pueden volverse obsoletos o eliminarse gradualmente. Esto puede incluir actualizaciones de UDI o etiquetado a medida que cambian las regulaciones.
P
Número de producto de farmacia (PPN)
La Agencia de Información de Productos Farmacéuticos (IFA»Informationsstelle für Arzneispezialitäten») emite el Código de la Agencia de Registro de Productos (PRA-Code) para cada sistema de numeración nacional que existe en el sector farmacéutico. El número de producto de farmacia (PPN) se introdujo con el fin de armonizar los diferentes sistemas de numeración, manteniendo al mismo tiempo los números de artículo nacionales para el etiquetado de envases y los sistemas de contabilidad en el sector sanitario. Sirve para cumplir con la Directiva 2011/62/UE para aumentar la seguridad contra la falsificación de medicamentos. El núcleo de este sistema es asignar un prefijo único, el código pra, a cualquier sistema de numeración. Para detectar errores en el número durante la entrada manual y el intercambio de datos, se añade un dígito de control de dos dígitos. El nuevo número se llama Número de Producto de Farmacia. Al número de producto de farmacia alemán (PZN «Pharmazentralnummer») se le asigna el código pra «11», el código pra austriaco «16». Esto permite mantener los sistemas nacionales de codificación existentes.
PI: Identificador de producción
Datos de producción (dinámicos) para un dispositivo, como la fecha de fabricación, la fecha de caducidad, el número de lote, el número de serie, etc.
Código PRA: Código de la agencia de registro de productos
Código asignado por el Centro de Información de Especialidades Farmacéuticas (IFA «Informationsstelle für Arzneispezialitäten») para cada sistema de numeración nacional en el campo de la farmacia. Forma parte del número de producto de farmacia (PPN).
Paquete de procedimientos (según MDR 2017/745)
En el Capítulo 1, Artículo 2, Párrafo 10 del MDR, el término Paquete de Procedimientos se explica de la siguiente manera:
«paquete de procedimientos»: una combinación de productos envasados juntos y comercializados con el fin de ser utilizados para un propósito médico específico;
PSR (Informe Resumido Periódico)
Informe simplificado que agrupa varios incidentes similares, en lugar de notificarlos individualmente como MIR.
PSRP (Informe Resumido Periódico – Actualización de Análisis Periódico)
Una actualización de un PSR existente cuando surgen nuevos hallazgos o datos. Ejemplo: análisis de seguimiento que muestra que la concentración de incidentes se mantiene estable o que las medidas correctivas están funcionando.
PSUR (Informe Periódico de Actualización de Seguridad)
Informe de seguridad periódico obligatorio según MDR/IVDR para dispositivos de clase IIa, IIb y III. Su contenido incluye un resumen de las actividades de PMS, datos de vigilancia, evaluación beneficio-riesgo, volúmenes de ventas y población usuaria.
PZN: Pharma Zentral Nummer
El Número Central de Farmacia (PZN) es una clave de identificación para medicamentos, dispositivos médicos y otros productos comúnmente utilizados en farmacias en toda Alemania. Es un número de ocho dígitos (7 dígitos + dígito de control) precedido por un signo menos, que identifica claramente los medicamentos por nombre, forma farmacéutica, concentración del ingrediente activo y tamaño del envase. Se imprime en texto plano (números) precedido por «PZN» y como código de barras (Código39) en cada paquete farmacéutico, por lo que la cadena de caracteres «PZN» no se incluye en el código de barras.
Q
SGC: Sistema de gestión de la calidad
Un sistema de gestión de la calidad es un método de gestión de la empresa. El objetivo es una gestión sistemática de la calidad.
QM: Gestión de la calidad
En la economía, la gestión de la calidad se refiere a una función (gestión) y a todas las medidas organizativas que sirven para mejorar la calidad del proceso, la calidad del trabajo y, por lo tanto, la calidad del producto y del servicio.
Código QR
El Código QR (Quick Response, como término de marca “QR Code”) es un código bidimensional (código de barras) desarrollado por la empresa japonesa Denso Wave en 1994. Debido a una corrección automática de errores, este método es muy robusto y, por lo tanto, ampliamente utilizado.
QS: Sistema de Calidad
Los fabricantes de dispositivos médicos deben establecer un sistema de calidad de acuerdo con la Administración de Alimentos y Medicamentos (FDA). Esto es para garantizar que sus productos siempre cumplan con los requisitos y especificaciones aplicables. Los sistemas de calidad para productos regulados por la FDA (alimentos, medicamentos, productos biológicos y dispositivos) se conocen como Buenas Prácticas de Manufactura Actuales (CGMP).
R
Servicio REST
La transferencia de estado representacional, REST para abreviar, es un paradigma de programación moderno para los servicios web. AWS, VMware, Azure y otros proveedores de nube hoy en día dependen casi exclusivamente de REST. Requiere menos recursos y es más fácil de usar. El servicio de transferencia de Europe IT Consultling GmbH para la transferencia de datos relevantes de UDI a la base de datos GUDID se basa en este método.
RFID
RFID o transpondedores son términos comunes para la tecnología de identificación por radiofrecuencia invisible. La identificación por radiofrecuencia «Identificación con la ayuda de ondas electromagnéticas» se refiere a un sistema transmisor-receptor para la identificación y localización automática y sin contacto de objetos y seres vivos utilizando ondas de radio.
Un sistema RFID consiste en un transpondedor que se encuentra sobre o en el objeto o criatura viva y contiene un código de identificación y un lector para leer este código.
S
SOX
Versión del software
Cualquier software que esté disponible comercialmente y, por lo tanto, represente un dispositivo médico independiente está sujeto a los requisitos de UDI. De este modo, la versión del software sirve como un elemento de identificación relevante, que se muestra en el UDI-PI.
Leer más
SRN: Número de registro único
El Número Único de Registro se utiliza para identificar de forma única a los operadores económicos en el contexto del Reglamento de Dispositivos Médicos o EUDAMED.
Estéril
Un material, un objeto, un líquido, una superficie o un entorno específico se describe como estéril (libre de gérmenes) si la cantidad de todos los microorganismos, así como virus, bacterias, priones y plásmidos se destruye o el residuo sobreviviente está por debajo de un cierto límite. La esterilidad es uno de los atributos que se deben tener en cuenta a la hora de recopilar datos relevantes de UDI.
Condiciones de almacenamiento y manipulación
Para EUDAMED, los requisitos de almacenamiento y manipulación que se requieren para el dispositivo. Por ejemplo: «no cortar», «almacenar en un recipiente cerrado», «evitar el contacto con el agua».
Para la FDA, estos se denominan «Tipos de almacenamiento y manipulación».
Para obtener más información, lea nuestro artículo Detalles sobre las condiciones de almacenamiento/manipulación y advertencias críticas
swissdamed
swissdamed es la base de datos suiza de productos sanitarios, gestionada por Swissmedic. Sirve para el registro de operadores económicos (CHRN) y de datos UDI de productos, y ofrece una función de búsqueda pública. Está concebida tomando como referencia a EUDAMED, pero funciona de manera independiente.
T
Tercero
Europe IT Consulting GmbH actúa como tercero en el contexto de la transmisión de datos a la FDA. Como proveedor de servicios externo, un tercero puede hacerse cargo de la transferencia de datos a la base de datos GUDID.
U
UDI: Identificación única del dispositivo
Sirve para la identificación única en todo el mundo de un dispositivo (médico).
UDI-DI y UDI-DI BÁSICO
Describe los atributos de exactamente un producto y tiene exactamente un BASIC-UDI.
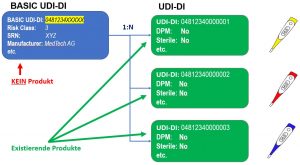
Unidad de uso DI
Una DI de unidad de uso es un identificador virtual asignado a un dispositivo médico individual cuando una UDI en el dispositivo individual no se identifica a nivel de su unidad de uso. Su propósito es asociar el uso de un dispositivo con/en un paciente cuando un paquete básico contiene más de un dispositivo.
V
Validación
En el contexto de la fabricación de dispositivos médicos y MDR, la validación es el proceso de confirmar que un proceso, sistema, material, método o producto cumple con los requisitos especificados. Esto es importante para garantizar que los dispositivos médicos sean seguros para su uso y funcionen según lo previsto.&#
Vigilancia
Este término se utiliza en el contexto del MDR y se refiere al proceso de monitoreo continuo de la seguridad de los dispositivos médicos que ya están en el mercado. El propósito de la vigilancia es identificar rápidamente cualquier problema o riesgo asociado con un dispositivo médico, lo que permite una acción correctiva oportuna si es necesario.
Verificación
En el contexto de UDI y MDR, la verificación es el proceso de verificar que un producto, sistema o componente cumpla con los requisitos especificados. Podría incluir la comprobación de que una etiqueta UDI se ha aplicado correctamente y se puede escanear con precisión, o que un dispositivo médico cumple con todos los requisitos reglamentarios.
Volumen de producción
Este término se refiere a la etapa de fabricación de dispositivos médicos en la que el dispositivo se produce en grandes cantidades. En esta etapa, es particularmente importante que todos los dispositivos se produzcan de manera consistente con el mismo estándar, y que los UDI se apliquen correctamente y se puedan leer correctamente.&
Proveedor
En el contexto de UDI, etiquetado y MDR, un proveedor es una empresa o individuo que vende productos o servicios a otra empresa. Desempeñan un papel fundamental en la cadena de suministro de dispositivos médicos y están sujetos a ciertos requisitos reglamentarios, incluida la necesidad de aplicar UDI a sus productos.
Impresión de datos variables (VDP)
Esta es una forma de impresión digital, incluida la impresión bajo demanda, en la que se pueden cambiar elementos como texto, gráficos e imágenes de una pieza impresa a otra, sin detener ni ralentizar el proceso de impresión. Se puede utilizar para imprimir etiquetas UDI con información única en cada etiqueta.
W
WebTrader
El FDA WebTrader es un portal web diseñado para remitentes de bajo volumen. WT permite a los usuarios iniciar sesión, firmar digitalmente los envíos y ver las respuestas a través de una interfaz web simple.
OMS: Organización Mundial de la Salud
La Organización Mundial de la Salud (OMS) es la autoridad coordinadora de las Naciones Unidas (ONU) para la salud pública internacional. Es un organismo especializado de las Naciones Unidas con sede en Ginebra. Fue fundada el 7 de abril de 1948 y ahora cuenta con 194 Estados miembros. Está encabezada por el Director General DE la OMS. Desde julio de 2017, está encabezada por el etíope Tedros Adhanom Ghebreyesus.
Enlace: https://www.who.int/
X
XML
El Extensible Markup Language, XML para abreviar, es un lenguaje de marcado para representar datos estructurados jerárquicamente en el formato de un archivo de texto que puede ser leído tanto por humanos como por máquinas.
Es utilizado por la FDA y EUDAMED como formato de intercambio de datos.
Y
SÍ ,
no hemos podido encontrar ningún término aquí. Tal vez puedas ayudarnos 😉
Z
CÓDIGO POSTAL
El formato de archivo ZIP es un formato para archivos comprimidos sin pérdidas, que por un lado reduce el espacio requerido para archivar y por otro lado funciona como un archivo contenedor en el que varios archivos relacionados o incluso árboles de directorios enteros zusammengefasst werden können.