
UDI terms and short-cuts in the medical industry from A-Z that you should know.
A B C D E F G H I J K L M N O P Q R S T U V W X Y Z
A
ACKs (Acknowledgments)
In the eMDR context, there are three acknowledgments: Ack1 confirms receipt of your submission at the ESG (Electronic Submissions Gateway), Ack2 indicates that the ESG successfully unpacked the file and forwarded it to CDRH, and Ack3 provides the processing result (accepted/error). The ESG is the FDA’s central submission portal (now ESG NextGen), and CDRH is the FDA’s Center for Devices and Radiological Health.
Adverse Event Reporting
This is the regulatory-mandated process by which manufacturers and operators capture, assess, and report adverse events (e.g., side effects, serious incidents) within required timeframes so authorities can detect safety signals and initiate actions such as recalls.
AIDC: Automatic Identification and Data Capture
AIDC systems can support the production, logistics, transport and distribution of goods, take over the tracking of goods and devices and check their marking and identification.
AIDC technologies include the graphical marking of goods with bar codes and 2D codes.
AIMDD
Active Implentable Medical Device Directive (will be replaced by MDR).
AS2 Protocol: Applicability Statement 2
The AS2 protocol is one of the most popular EDI (Electronic Data Interchange) transmission standards. The reason for its widespread use in business is that AS2 uses the Internet as a communication channel. Therefore the protocol does not depend on the use of a proprietary network. For transmission over the Internet, AS2 uses both HTTP and HTTPS protocols.
AS4 Protocol: Applicability Statement 4
The AS4 Protocol is a Web services based messaging protocol for the secure exchange of business-to-business messages between trading partners. The protocol was approved by the technical committee of OASIS (Organization for the Advancement of Structured Information Standards) to ebXML Messaging Services. With its web services capability, AS4 has the chance to become the cloud-based communication standard.
AusUDID
The Australian Unique Device Identification Database (AusUDID) is the Therapeutic Goods Administration’s (TGA) public UDI database.
Authorised Representative
An authorized representative is a representative who acts and decides on behalf of another person by means of a declaration of intent or by receiving it.
The definition, rights and duties of an authorised representative are described in chapter 11 of the MDR.
Read more about >>MDR Authorized Representative<< Read more >>UDI Roles<<B
Basic UDI-DI Code
The BASIC UDI-DI summarizes the common characteristics of a group of products.
Budi
Is used to shorten BASIC UDI-DI.
C
CA: Competente Authority
Responsible agency for the approval of medical devices.
Read moreCHRN
Unique registration number issued by Swissmedic for economic operators.
Clinical Investigation
Defined as “any systematic investigation or study in or on one or more human subjects, undertaken to assess the safety and/or performance of a medical device.” (SG5/N1:2007).
It assesses the safety and clinical performance of the device in question and evaluate whether the device is suitable for the purpose(s) and the population(s) for which it is intended (EN ISO 14155-1:2009).
Read chapter VI of the MDR for more information about clinical investigation concerning devices placed in the EU.
CND: Classificazione Nazionale dei Dispositivi medici
The Italian system, in English “National Classification of Medical Devices” has applied for registration of medical devices at EUDAMED. There it is called EMDN.
Code 128
The Code128 is an alphanumeric barcode with high information density. The principle structure consists of a start character, the user information, the check digit and the stop character. A white area (bright field or quiet zone) must be kept free before the start character and after the stop character.
Critical warnings
Warnings or precautions to be taken that need to be brought to the immediate attention of the user of the device, and to any other person. This information may be kept to a minimum in which case more detailed information shall appear in the instructions for use, taking into account the intended users. (Source: Medical device Regulation 2017/745, Annex I, 23.2 about the information the label shall bear)
For example: ” if swallowed: get immediate medical advice/attention”, “explosive when dry”, “toxic by eye contact”.
For more information, read our article Details on Storage/Handling conditions and Critical warnings
CSDT: Common Submission Dossier Template
ASEAN document intended to provide a common template for the submission of medical device information to medical device regulatory authorities of ASEAN member countries. Here is the ASEAN guidance about this template.
Custom-Made Device
– device specifically made in accordance with a written prescription of any person authorised by national law by virtue of that person’s professional qualifications
– device which gives specific design characteristics provided under that person’s responsibility
– device intended for the sole use of a particular patient exclusively to meet their individual conditions and needs.
– As long as the product is not “mass-produced” (manufactured in large quantities by an automated mechanical process), manufacturers may use modern state of the art technologies to manufacture CMDs.
For more information, read our dedicated article on this topic: Custom-made Device (CMD) according to MDR (2017/745).
D
DataMatrix Barcode
The DataMatrix code is one of the best known 2D codes. It is used for permanent direct marking, but also increasingly as printed code image in document handling.
Dimdi
The German Institute for Medical Documentation and Information was a subordinate authority of the Federal Ministry of Health.
Distributor (According to MDR)
The definition, rights and obligations of distributors are described in chapter 14 of the MDR.
Read moreDI: Device Identifier
Product identification data (static) for a device such as GTIN.
DM: Direct Marking
A device identification number. For example GTIN14 number or HIBC number.
DPM: Direct Part Marking
Marking of a product without label. For example laser engraving of a serial number on surgical instruments.
DUNS: Data Universal Numbering System
The DUNS number is a numerical code developed by Dun&Bradstreet and used internationally as a standard for the unique identification of companies.
E
EMDN
The European Nomenclature on Medical Devices has established itself as the system for registration at EUDAMED.
eMDR
Refers to the electronic submission of Medical Device Reports (MDRs) to the U.S. FDA by manufacturers, importers, and user facilities.
ESG: Electronic Submissions Gateway
The Electronic Submissions Gateway of the Food and Drug Administration is an agency-wide solution for the acceptance of electronic submissions. The FDA ESG is a highly scalable, readily available, powerful and secure exchange point for the FDA and its partners to handle a variety of documents and submissions via industry standard protocols. The FDA ESG enables the secure submission of pre- and post-market regulatory information for review.
The FDA ESG offers two methods, WebTrader and AS2, for submitting applications to the FDA.
EUDAMED: European Database for Medical Devices
Database for registration of medical devices in the European Union. A new version of the database will be made available as part of the introduction of the new Medical Device Regulation.
EURL: EU Refrence Leboratory
Laboratories designated by the European Commission to deliver opinions and views for some high-risk and IVD medical devices and have an advisory role to actors such as the European Commission or the MDCG. The tasks of the EURLS are defined in the Article 100 paragraph 2 and 3 of the In Vitro Diagnostic Medical device Regulation.
F
FDA
The U. S. FDA is the food and drug administration of the United States. As such, it reports to the U.S. Department of Health and Human Services.
Link: https://www.fda.gov/
FSCA: Field Safety Corrective Action
Any action taken by a device owner to reduce the risk of death or serious deterioration of health associated with the use of a medical device already on the market.
Regarding FSCA in the EUDAMED, please refer to Article 87 of the MDR and Article 82 of the IVDR.
FSN (Field Safety Notice)
ccompanying document for an FSCA – the notification sent to customers/users. It describes the issue, the affected products, and provides instructions (e.g., stop using the product).
G
GDPR
The General Data Protection Regulation is a data protection regulation introduced in the European Union (EU). It came into effect on May 25, 2018, with the aim of strengthening and harmonizing the protection of personal data of EU citizens. The GDPR establishes strict rules on how personal data may be collected, processed, stored, and transferred.
Link: https://gdpr-info.eu/
GDSN
The Global Data Synchronization Network (GDSN) is an Internet-based system for data transmission used worldwide. GDSN ensures that the data exchanged between trading partners complies with GS1 standards worldwide.
Link: https://www.gs1.org/services/gdsn
GHTF: Global Harmonization Task Force on Medical Devices
The Global Harmonization Task Force was “a voluntary group of representatives of the national regulatory authorities for medical devices” whose goal was the worldwide standardization of the regulation of medical devices. It consisted of members from industry.
The GHTF disbanded at the end of 2012. Its role was taken over by the International Medical Device Regulators Forum (IMDRF), a follow-up organization composed of officials from regulatory authorities – not industry – from around the world. The GHTF website is no longer functional.
GMDN: Global Medical Device Nomenclature
The GMDN code is required to register medical devices on the FDA database named GUDID.
Link: https://www.gmdnagency.org/
GS1
GS1 is a network of non-profit organisations that develop, negotiate and maintain standards for cross-company processes worldwide. They have been appointed as the allocation body for the operation of a system for the allocation of UDI in the context of the unique product identification of medical devices.
Link: https://www.gs1.org/
GTIN
The Global Trade Item Number is an identification number derived from the GS1 system (formerly EAN system) with which trading units can be marked.
GUDID
The Global Unique Device Identification Database is a database managed by the FDA that serves as a reference catalog for each device with a unique device identifier (UDI).
Link to GUDID Productiv: https://gudid.fda.gov/gudid/app/login.xhtml
Link to GUDID PreProd: https://gudid.preprod.fda.gov/gudid/
H
HIBC: Health Industry Barcode
Worldwide unique number which serves for the identification of products.
Link: https://www.hibcc.org/
HIPAA
Link: https://www.cdc.gov/phlp/publications/topic/hipaa.html
HL7
Health Level 7 is a group of international standards for the exchange of data between healthcare organisations and their computer systems.
Read moreI
ICCBBA: International Council for Commonality in Blood Banking Automation
The ICCBBA is an international, non-profit, non-governmental standardisation organisation that has an official relationship with the World Health Organisation (WHO) and is responsible for the management and development of the ISBT 128 standard. It manages, develops and licenses the international information standard for the terminology, coding and labelling of medical devices of human origin. The ICCBBA acts as an issuing agency in the field of UDI.
Link: https://www.iccbba.org/
ICSR Report
An ICSR (Individual Case Safety Report) is a standardized safety report on a single case of an adverse event or product problem. In drug and medical device vigilance (e.g., under ICH E2B(R3)), it is exchanged electronically to ensure that cases are reported and evaluated consistently.
IFA – Informationsstelle für Arzneispezialitäten
“Information centre for pharmaceutical specialities”
IFA GmbH was designated as the allocation body for UDI-DI by the EU Commission in its implementing decision of 6 June 2019. A Pharmacy Product Number (PPN) encoded on the package according to the IFA coding system thus represents an MDR-compliant UDI-DI.
Link: http://www.ifaffm.de/en/home.html
IMDRF: International Medical Device Regulators Forum
The International Medical Device Regulators Forum was conceived in February 2011, as a forum to identify future guidelines for the harmonisation of medical device regulations.
It is a voluntary group of medical device regulators from around the world who have come together to build on the strong groundwork of the Global Harmonization Task Force on Medical Devices (GHTF) and to accelerate the international harmonization and convergence of medical device regulation.
Link: http://imdrf.org/
Importers
The definition, rights and obligations of importers are described in chapter 13 of the MDR.
Read moreIssuing Agency
The Issuing Agency is responsible for the operation of a system for the allocation of UDI within the framework of the unique device identification for medical devices. Well-known issuing agencies are GS1, HIBCC and others.
Read moreIVD: In-vitro-Diagnostic
In vitro diagnostic medical device is a term for medical devices used for medical laboratory examination of samples taken from the human body. These are examined outside the body (Latin in vitro ‘in glass’).
IvDD
In Vitro Diagnostics Directive (to be replaced by IVDR).
IVDR: In Vitro Diagnostica Regulation
The new European Regulation on in vitro diagnostic medical devices replaces the existing In Vitro Diagnostics Directive.
It is valid since 25.05.2017.
J
JSON: JavaScript Object Notation
The JavaScript Object Notation is a compact data format in an easy to read text form and serves the purpose of data exchange between applications. JSON is independent of the programming language. Parsers and generators exist in all common languages.
K
Kit (EU)
The kit is described in the legal framework of the European Union only in the IVDR.
‘kit’ means a set of components that are packaged together and intended to be used to perform a specific in vitro diagnostic examination, or a part thereof
Quelle: REGULATION (EU) 2017/746, Chapter 1, Article 2, Definitions (Paragraph 11)
Kit (USA)
In the FDA’s definition, the kit or conveniece kit should be interpreted as consisting of two or more different medical devices packaged together for the convenience of the user.
The original definition can be found under the following Link
L
Latex
The material latex is contained in many products and must always be declared. It can cause a strong allergic reaction in some people and damage their health. This is one of the reasons why this attribute must always be declared when registering medical products.
Legacy Device
Medical Devices, Active Implantable Medical Devices and In Vitro Diagnostic Medical Devices that are covered by a valid certificate issued in accordance with Directive 93/42/EEC, Directive 90/385/EEC or Directive 98/79/EC and that continue to be placed on the market after the date of application of Regulation (EU) 2017/745 (MDR) or Regulation 2017/746 (IVDR). EU Guidance here.
Legislation
The European legislations which are in the focus of UDI are:
Medical Device Regulation (MDR), In-Vitro Diagnostic Regulation (IVDR), Medical Device Directive (MDD), Active Implantable Medical Device Directive (AIMDD), In-Vitro Diagnistic Directive (IVDD)
M
MASTER-UDI
The European Commission is amending Regulation (EU) 2017/745 to improve the Unique Device Identification (UDI) system for contact lenses and in the later stage for products which can have many variations.
The current system requires manufacturers to assign a UDI to each variant of contact lenses, which has led to an overwhelming number of UDI-DIs (Device Identifiers) in the European Database on Medical Devices (Eudamed).
To address this issue, the Commission has developed the “Master UDI-DI” concept, which groups contact lenses with the same clinical and design parameter combinations under one identifier. This amendment will make the identification process more efficient, without compromising safety or traceability.
The new regulation will apply two years after its entry into force, but manufacturers may start using the Master UDI-DI and UDI-PI (Production Identifier) earlier if desired.
You can find more information here: EUDAMED Master-UDI
Manufacturer (According to MDR)
The definition, rights and obligations of manufacturers are explained in chapter 10 of the MDR.
Read moreMDCG: Medical Device Coordination Group
The Medical Device Coordination Group advises the European Commission and supports the Commission and the Member States in ensuring a harmonised implementation of the Medical Device Regulations (EU) 2017/745 and 2017/746.
MDD
Medical Device Directive (will be replaced by MDR).
MDR: Medical Device Regulation
The new European Medical Devices Regulation replaces the existing medical devices directives.
It is valid since 25.05.2017.
MDSAP: Medical Device Single Audit Program
It allows an MDSAP recognized Auditing Organization to conduct a single regulatory audit of a medical device manufacturer that satisfies the relevant requirements of the regulatory authorities participating in the program.
The members are: Australia, Brazil, Canada, Japan, FDA. the EU, UK, and WHO are only official observers.
MIR (Manufacturer Incident Report)
Report of a serious incident (death, serious deterioration of health, or risk to public health).
M2M: Machine-to-Machine
Machine-to-machine stands for the automated exchange of information between terminal devices such as machines, vending machines, vehicles or containers with each other or with a central control centre, increasingly using the Internet and the various access networks, such as the mobile phone network.
MTR (Manufacturer’s Trend Report)
Report submitted to the authority when the manufacturer identifies a noticeable increase (trend) in non-serious incidents or expected side effects.
N
NANDO
New Approach Notified and Designated Organisations (Notified Body, for example TÜV Süd).
Link: https://ec.europa.eu/growth/tools-databases/nando/
NDC: National Drug Code
The National Drug Code (NDC) is a unique product identifier used in the United States for drugs intended for human use. The Drug Listing Act of 1972 requires registered drug establishments to provide the Food and Drug Administration (FDA) with a current list of all drugs manufactured, prepared, propagated, compounded, or processed by it for commercial distribution. Drug products are identified and reported using the NDC.
Notified Body
The Notified Bodies of the European Union are private inspection bodies (auditing and certification bodies) notified and supervised by the state, which act on behalf of manufacturers in order to accompany and control the conformity assessment of manufacturers of industrial products of various kinds.
Read more
O
Optical Character Recognition (OCR)
OCR is a technology used to convert different types of documents, such as scanned paper documents, PDF files or images captured by a digital camera, into editable and searchable data. In the context of UDI and labeling, OCR might be used to capture and digitalize information from labels.
Owner/Operator
In the U.S. FDA UDI regulations, this term refers to the entity responsible for manufacturing, repackaging, relabeling, and distributing a device.
Off-label use
This refers to the use of a medical device (or drug) for an unapproved indication or in an unapproved age group, dosage, or route of administration. This concept may come up in discussions about labeling, as labels should accurately reflect the approved uses of a product.
OEM (Original Equipment Manufacturer)
In the context of medical devices, an OEM is a company that produces parts and equipment that may be marketed by another manufacturer. The OEM may also be responsible for certain aspects of UDI and labeling, depending on regulatory requirements.
Obsolescence Management
This refers to the practice of ensuring that a device remains compliant with current regulations throughout its lifecycle, even as some components or materials used in its construction may become obsolete or be phased out. This can include updates to UDI or labeling as regulations change.
P
Pharmacy Product Number (PPN)
The Information Agency for pharmaceutical Products (IFA”Informationsstelle für Arzneispezialitäten”) issues the Product Registration Agency Code (PRA-Code) for each national numbering system that exists in the pharmaceutical sector. The Pharmacy Product Number (PPN) was introduced in order to harmonise different numbering systems while retaining the national article numbers for packaging labelling and accounting systems in the health care sector. It serves to comply with Directive 2011/62/EU to increase the anti-counterfeiting security of medicinal products. The core of this system is to assign a unique prefix, the PRA code, to any numbering system. In order to detect errors in the number during manual input and data exchange, a two-digit check digit is added. The new number is called Pharmacy Product Number. The German Pharmacy Product Number (PZN “Pharmazentralnummer”) is assigned the PRA code “11”, the Austrian PRA code “16”. This allows existing national coding systems to be maintained.
PI: Production Identifier
Production data (dynamic) for a device such as date of manufacture, expiry date, batch number, serial number, and so on.
PRA-Code: Product Registration Agency Code
Code assigned by the Information Centre for Proprietary Medicinal Products (IFA “Informationsstelle für Arzneispezialitäten”) for each national numbering system in the field of pharmacy. It is part of the Pharmacy Product Number (PPN).
Procedure Pack (according to MDR 2017/745)
In Chapter 1, Article 2, Paragraph 10 of the MDR, the term Procedure Pack is explained as follows:
‘procedure pack’ means a combination of products packaged together and placed on the market with the purpose of being used for a specific medical purpose
PSR (Periodic Summary Report)
Simplified collective report for multiple similar incidents, instead of reporting each one individually as an MIR.
PSRP (Periodic Summary Report – Periodic Analysis Update)
An update to an existing PSR when new findings or data are available. Example: follow-up analysis showing that the clustering of incidents remains stable or that corrective measures are effective.
PSUR (Periodic Safety Update Report)
Periodic safety report required under MDR/IVDR for Class IIa, IIb, and III devices. Contents include a summary of PMS activities, vigilance data, benefit-risk evaluation, sales volumes, and user population.
PZN: Pharma Zentral Nummer
The Pharmacy Central Number (PZN) is an identification key for drugs, medical devices and other products commonly used in pharmacies throughout Germany. It is an eight-digit number (7 digits + check digit) preceded by a minus sign, which clearly identifies medicinal products by name, dosage form, strength of active ingredient and package size. It is printed in plain text (numbers) preceded by “PZN” and as a bar code (Code39) on each pharmaceutical package, whereby the character string “PZN” is not included in the bar code.
Q
QMS: Quality management system
A quality management system is a method of company management. The aim is a systematic quality management.
QM: Quality management
In the economy, quality management refers to a function (management) and all organisational measures that serve to improve process quality, work quality and thus product and service quality.
QR-Code
The QR Code (Quick Response, as a trademark term “QR Code”) is a two-dimensional code (barcode) developed by the Japanese company Denso Wave in 1994. Due to an automatic error correction this method is very robust and therefore widely used.
QS: Quality System
Medical device manufacturers must establish a quality system according to the Food and Drug Administration (FDA). This is to ensure that their products always meet the applicable requirements and specifications. The quality systems for products regulated by the FDA (food, drugs, biological products and devices) are known as Current Good Manufacturing Practices (CGMPs).
R
REST Service
Representational State Transfer, REST for short, is a modern programming paradigm for Web services. AWS, VMware, Azure and other cloud providers today rely almost exclusively on REST. It requires fewer resources and is easier to use. The transfer service of Europe IT Consultling GmbH for the transfer of UDI relevant data to the GUDID database is based on this method.
RFID
RFID or transponders are common terms for the invisible radio frequency identification technology. Radio-frequency identification “Identification with the aid of electromagnetic waves” refers to a transmitter-receiver system for automatic and contactless identification and localisation of objects and living beings using radio waves.
An RFID system consists of a transponder which is located on or in the object or living creature and contains an identifying code, and a reader for reading this code.
S
SOX
Software Version
Any software that is commercially available and thus represents a stand-alone medical device is subject to UDI requirements. Thereby, the software version serves as a relevant identification element, which is displayed in the UDI-PI.
Read moreSRN: Single Registration Number
The Single Registration Number is used to uniquely identify economic operators in the context of the Medical Device Regulation or EUDAMED.
Sterile
A material, an object, a liquid, a surface or a specific environment is described as sterile (germ-free) if the quantity of all microorganisms as well as viruses, bacteria, prions and plasmids is killed or the surviving residue is below a certain limit. Sterility is one of the attributes that must be taken into account when collecting UDI relevant data.
Storage and Handling Conditions
For EUDAMED, storage and handling requirements that are required for the device. For example: “do not cut”, “store in a closed container”, “avoid contact with water”.
For the FDA, these are called “Storage and Handling Types”.
For more information, read our article Details on Storage/Handling conditions and Critical warnings
swissdamed
swissdamed is Switzerland’s medical device database, operated by Swissmedic. It is used for the registration of economic operators (CHRN) and UDI product data, and it provides a public search function. It is modeled after EUDAMED but operates independently.
T
Third Party
Europe IT Consulting GmbH acts as a third party in the context of data transmission to the FDA. As an external service provider, a third party can take over the data transfer to the GUDID database.
U
UDI: Unique Device Identification
Serves for the worldwide unique identification of a (medical) device.
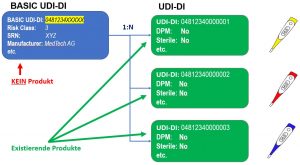
UDI-DI and BASIC UDI-DI
Describes the attributes of exactly one product and has exactly one BASIC-UDI.
Unit of Use DI
A Unit of Use DI is a virtual identifier assigned to an individual medical device when a UDI on the individual device is not identified at the level of its Unit of Use. Its purpose is to associate the use of a device with/on a patient when a basic package contains more than one device.
V
Validation
In the context of medical device manufacturing and MDR, validation is the process of confirming that a process, system, material, method, or product meets the specified requirements. This is important to ensure that the medical devices are safe for use and perform as intended.
Vigilance
This term is used in the context of the MDR and refers to the process of continuous monitoring of the safety of medical devices that are already on the market. The purpose of vigilance is to swiftly identify any issues or risks associated with a medical device, allowing for timely corrective action if necessary.
Verification
In the context of UDI and MDR, verification is the process of checking that a product, system, or component meets specified requirements. It could include checking that a UDI label has been correctly applied and can be accurately scanned, or that a medical device meets all regulatory requirements.
Volume Production
This term refers to the stage of medical device manufacturing where the device is produced in large quantities. At this stage, it’s particularly important that all devices are consistently produced to the same standard, and that UDIs are correctly applied and can be read correctly.
Vendor
In the context of UDI, labeling, and MDR, a vendor is a company or individual that sells products or services to another company. They play a critical role in the supply chain of medical devices, and are subject to certain regulatory requirements, including the need to apply UDIs to their products.
Variable Data Printing (VDP)
This is a form of digital printing, including on-demand printing, in which elements such as text, graphics, and images may be changed from one printed piece to the next, without stopping or slowing down the printing process. It can be used for printing UDI labels with unique information on each label.
W
WebTrader
The FDA WebTrader is a web portal designed for low volume submitters. WT allows users to log in, digitally sign submissions and view responses through a simple web interface.
WHO: World Health Organization
The World Health Organization (WHO) is the United Nations (UN) coordinating authority for international public health. It is a specialised agency of the United Nations with headquarters in Geneva. It was founded on 7 April 1948 and now has 194 member states. It is headed by the WHO Director-General. Since July 2017, it has been headed by the Ethiopian Tedros Adhanom Ghebreyesus.
Link: https://www.who.int/
X
XML
The Extensible Markup Language, XML for short, is a markup language for representing hierarchically structured data in the format of a text file that can be read by both humans and machines.
It is used by the FDA and EUDAMED as a data exchange format.
Y
YES ,
we couldn’t find any terms here. May be you can help us 😉
Z
ZIP
The ZIP file format is a format for losslessly compressed files, which on the one hand reduces the space required for archiving and on the other hand functions as a container file in which several related files or even entire directory trees zusammengefasst werden können.