Preguntas y respuestas de EUDAMED
Índice
Actores y roles en EUDAMED
Registro del producto – Principios y plazos
- Plazos: ¿Cuándo será obligatorio EUDAMED?
- Obligación de registro para los productos de clase I
- Registro UDI obligatorio para los productos IVD
- Obligación de registro para los productos de inventario IVDD
Registro del producto – Práctica y ejecución
- Registro de un producto individual – paso a paso
- Así funciona el registro de productos en EUDAMED
- Editar productos ya registrados
- Cambios en el producto (nombre, nombre comercial, etc.)
- Advertencias y contraindicaciones
- Modificar posteriormente el UDI-DI básico
Estructura de datos y datos obligatorios
- Modelo del dispositivo: ¿qué introducir?
- Datos obligatorios para el registro del producto
- Información de mercado – Campos de fecha
- Lista de países: ¿Se aplica solo a la UE?
- "Originalmente comercializado" – significado
Agrupación y codificación
- Agrupar productos bajo un UDI-DI básico
- Códigos GS1 para dispositivos heredados
- Falta el código EMDN, ¿qué hacer?
GUI y funciones del sistema
- Funciones en la interfaz gráfica de EUDAMED
- Carga de Excel y prevención de duplicación de mantenimiento de datos
- Recomendación de herramientas para el rodillo del fabricante
Sistemas nacionales vs. EUDAMED
Pregunta: ¿Puede una empresa registrada como importador en EUDAMED asociarse con un fabricante registrado en EUDAMED sin que se envíe una notificación al fabricante y se obtenga su aprobación?
No, esto no es posible sin el conocimiento y la participación activa del fabricante .
En EUDAMED, la vinculación de los actores -por ejemplo, el importador con el fabricante- es siempre un proceso recíprocoque requiere la confirmación o aprobación del otro socio .En detalle:
El importador puede iniciar la vinculaciónindicando en su cuenta EUDAMED (en el módulo Actor) que está vinculado a un fabricante concreto (indicando su SRN).
El fabricante debe aceptar explícitamente esta solicitudde enlace. Sin la aprobación del fabricante, el enlace no se activa.
Pregunta: La mayoría de nuestros productos caen bajo el rollo del fabricante, pero hay algunos productos que traemos al mercado con el rollo de importación. Para el rollo de importación, me gustaría saber cómo se realizará el registro.
Registro como importador en EUDAMED – paso a paso
1. Comprender el rollo de importación (según MDR, artículo 13):
Un importador es una empresa con sede en la UEque introduce un producto en el mercado de la UE desde fuera de la UE , es decir, por ejemplo, primero importa y distribuye productos de un fabricante de fuera de la UEa la UE.
Importante:
Solo es necesario si sus productos son importados de un fabricante no perteneciente a la UE.
Si usted mismo es un fabricante con sede en la UE y vende sus propios productos, no es un importador en el sentido del MDR.
📝 ¿Cómo registrarse como importador?
A. Paso 1: Registro en el módulo Actor
Seleccione "Importador" como rol de actor.
Presente el registro del actor ante su autoridad nacional (en DE: BfArM, en FR: ANSM, etc.).
Después de la aprobación, recibirá su SRN (número de registro único) como importador.
📌 Si ya está registrado como fabricante, también debe realizar un segundo registro como importador, con un rol separado, pero posiblemente la misma empresa.
B. Paso 2: Vinculación con fabricantes no pertenecientes a la UE
Una vez que tenga un SRN como importador, puede conectarse con los respectivos fabricantes nopertenecientes a la UE cuyos productos importe a la UE.
La vinculación se realiza de forma recíproca : el fabricante debe dar su consentimiento.
Esta vinculación es un requisito previo para que los productos en cuestión se asignen correctamente en EUDAMED.
📌 Obligaciones importantes en el rollo de importación (MDR artículo 13):
Tarea Descripción Asegurar el cumplimiento de MDR El importador debe comprobar si el producto tiene el marcado CE, si hay instrucciones de uso, etc. Comprobar la conformidad con el UDI El embalaje debe llevar correctamente la etiqueta UDI Asegurar la trazabilidad del producto El importador debe documentar su cadena de suministro Comprobar entradas UDI/Device en EUDAMED Los productos importados deben estar debidamente registrados Cooperación con las autoridades El importador debe poder informar de retiradas, incidentes, etc.
🧠 Característica especial: Combinación de fabricante e importador
Por ejemplo, si fabrica sus propios productos (rol de fabricante) e importa productos adicionales de un fabricante de fuera de la UE bajo su propio SRN, entonces:
debe registrarse dos veces: una como fabricante y otra como importador
debe cumplir con las obligaciones del importador por separado para los productos importados
no debe registrar los mismos productos como fabricante e importador al mismo tiempo ; debe estar claramente asignado
📋 Nota adicional:
El registro del producto es responsabilidad del fabricante no perteneciente a la UE para los productos importados (módulo de dispositivo).
El importador está vinculado en el sistema como operador económico, no como registrador principal.
Pregunta: ¿Cuál es la delimitación clara de los roles en EUDAMED?
Delimitación clara de los roles en EUDAMED: ¿quién hace qué?
En EUDAMED hay 6 roles de actor ,pero para el registro del producto y las obligaciones, estos 4 son los más importantes:
Rol ¿Requiere SRN? ¿Es posible registrar el producto? Tareas típicas Fabricante (Manufacturer) ✅ Sí ✅ Sí Registra productos (UDI), gestiona datos de dispositivos, vigilancia Representante autorizado ✅ Sí No (excepto si es delegado por el fabricante) Representa a fabricantes no pertenecientes a la UE, puede asumir tareas administrativas Importador (Importador) ✅ Sí ❌ No Se vincula con el fabricante, asume las obligaciones de MDR (por ejemplo, inspección de etiquetas, trazabilidad) Fabricante del paquete del sistema/procedimiento ✅ Sí ✅ Sí Informa de conjuntos que constan de varios productos 🔍 1. El papel del "fabricante" (Manufacturer)
Tiene control total sobre el registro del producto
Debe registrar cada Basic UDI-DI y UDI-DI
Puede ser fabricante tanto de la UE como de fuera de la UE (no perteneciente a la UE con mandato de los representantes autorizados de la UE)
Vincula productos con operadores económicos (importadores, distribuidores)
Responsable de notificaciones devigilancia, tecn. Documentación, destino, etc.
🔍 2. El papel del "importador"
Solo se puede registrar si la sede de la empresa se encuentra dentro de la UE
No registra ningún producto por sí mismo , pero debe:
Vincularse con el fabricante
Asegurarsede que los productos que importa estén registrados en EUDAMED
Hacer visible su propia empresa en el módulo Economic Operator
El importador debe poner su SRN en la etiqueta (art. 13 MDR)
📌 Una misma empresa puede ser tanto fabricante como importador, pero en EUDAMED son dos funciones separadas, cada una con su propio SRN.
🔍 3. El papel de "Representante autorizado"
Necesario si el fabricante no está establecido en la UE
Se vincula con el fabricante no perteneciente a la UE mediante una declaración de mandato
No registre productos usted mismoa menos que asuma esta tarea en nombre del fabricante
👥 Vinculación de roles – ejemplo de la práctica
Así pues, si:
Elfabricante tiene su sede en la UE → Vosotros mismos registráis los productos (datos UDI, etc.)
Al mismo tiempo, importador de productos de un fabricante de EE. UU. → Se registra por separado como importador, obtiene su propio SRN y se vincula con el fabricante de EE. UU.
📄 En resumen: ¿qué significa esto para vosotros?
Si Uds…. Entonces tenéis que… fabrica sus propios productos en la UE Regístrese como fabricante , registre los productos usted mismo Productos importados de terceros países a la UE Además, regístrese como importador y vincúlelo con el fabricante actúa para fabricantes no pertenecientes a la UE Actuar como apoderado o garantizar una conexión contractual
🎯 Conclusión:
EUDAMED está estructurado con precisión, y cada rol tiene sus propias tareas, deberes y su propio SRN. Una vez que una empresa asume varios roles, necesita procesos internos claros y asignación desistemas para evitar confusiones o problemas de auditoría.
Pregunta: Plazos: La Comisión Europea ha anunciado que el módulo de registro UDI pronto estará completamente operativo, con la fecha límite para el registro de los productos a principios del próximo año. ¿Se espera que se cumpla este plazo y que EUDAMED esté operativo a tiempo?
Versión: Q1/2025:
Sí, se esperaque la Comisión de la UE confirme oficialmente el funcionamiento completo del módulo EUDAMED-UDI/Device en el primer semestre de 2025 en el Diario Oficial de la UE (OJEU) . Se espera que el plazo para el registro obligatorio del producto sea de 24 meses después de este anuncio .
👉 Esto significa que el plazo finalizará como muy pronto a principios de 2027.
📅 ¿Cuál es el estado actual (marzo de 2025)?
El módulo UDI/Device ya es completamente funcional desde el punto de vista técnico y se puede utilizar de forma voluntaria.
La Comisión de la UE está llevando a cabo las últimas pruebas de interoperabilidad (M2M) con los operadores económicos y las autoridades nacionales.
El anuncio oficial en el Diario Oficial de la UE (OJEU) aún está pendiente: marca el "pistoletazo de salida" para el período de transición de 24 meses.
🧠 ¿Qué significa esto para los fabricantes e importadores?
Tema Significado Uso voluntario Ahora ya es posible y recomendado: ayuda con la seguridad interna del proceso y el mantenimiento de datos Uso obligatorio No inicia ninguna obligación legal hasta → después del anuncio de OJEU Obligación de registro UDI Se requieren 24 meses después de la publicación de OJEU para todos los productos MDR Recomendación Empieza a registrarte ahora para evitar cuellos de botella y errores 📌 Consejo práctico:
Muchas empresas subestiman el esfuerzo necesario para mantener los datos UDI, especialmente en el caso de grandes carteras. Quien espere hasta el compromiso corre el riesgo de no realizar el esfuerzo durante el período de transición o de tener problemas en las auditorías.
💬 Conclusión:
Se espera que el plazo se publique en el Diario Oficial, probablemente en el transcurso de 2025.
La obligación de registro es válida a partir de entonces + 24 meses, es decir, probablemente a partir de principios de 2027.
Técnicamente, EUDAMED está listo : las empresas deben prepararse ahora, incluso si la obligación aún no es efectiva.
Pregunta: ¿Cuáles son los plazos para el registro de productos de clase I?
La obligación de registro para los productos de clase I en EUDAMED no comenzará hasta que la Comisión de la UE publique oficialmente la funcionalidad completa de EUDAMED en el Diario Oficial (OJEU). A partir de ese momento se aplicará un periodo transitorio de 6 meses.
📅 Esto significa concretamente:
Paso Fecha Publicación OJEU (función EUDAMED completa) ⚠️ Pendiente (previsto para 2025) Inicio de la obligación de registro del producto (clase I) 📆 6 meses después de la publicación de OJEU El registro debe completarse 📆 Se espera que comience/mediados de 2026, dependiendo de la fecha de publicación
Pregunta: ¿ Tengo que utilizar ya EUDAMED para el etiquetado UDI (según IVDR) o desde cuándo es necesario?
No, actualmente no existe ninguna obligación legal de registrar datos UDI en EUDAMED, ni siquiera bajo IVDR.
SIN EMBARGO,
se espera que la Comisión de la UE anuncie la funcionalidad completa de EUDAMED (incluido el módulo UDI) en el Diario Oficial de la UE en el transcurso de 2025 (OJEU).
👉 A partir de ese momento, transcurrirá un periodo transitorio de 24 meses.Esto significa que el registro
UDIen EUDAMED será obligatorio a partir de 2027 como muy pronto, también para los productos IVD.
Pregunta: ¿Los productos existentes de acuerdo con IVDD también deben almacenarse allí?
No, los productos existentes que todavía se comercializan bajo el IVDD no tienen que registrarse actualmente en EUDAMED.
La obligación de registro UDI solo se aplica a los productos compatibles con IVDR.Período transitorio y excepciones
🧾 ¿Qué son los "productos existentes"?
Estos son productos que han sido certificados de acuerdo con IVDD y pueden seguir comercializándose de acuerdo con los períodos transitorios del artículo 110 del IVDR , por ejemplo, sobre la base de un antiguo certificado de cuerpo notificado. Dependiendo de la clase, dichos productos pueden seguir comercializándose a más tardar en mayo de 2026 o mayo de 2027 , sin que ya deban cumplir con el IVDR.
❗ Para estos productos se aplica lo siguiente:
Aún no necesita la identificación UDI
No es necesario estar registrado en EUDAMED
No se crean nuevas obligaciones en EUDAMED , ni para el módulo de dispositivo ni para la vigilancia
🚦 ¿Cuándo deben registrarse los productos IVD en EUDAMED?
Tipo de producto ¿Es necesario registrarse en EUDAMED? Nuevo producto compatible con IVDR ✅ Sí, después del inicio de la obligación de UDI (esperado a partir de 2027) Producto existente según IVDD ❌ No, siempre y cuando se revenda por regla transitoria Producto reconvertido (recertificación IVDR) ✅ Sí
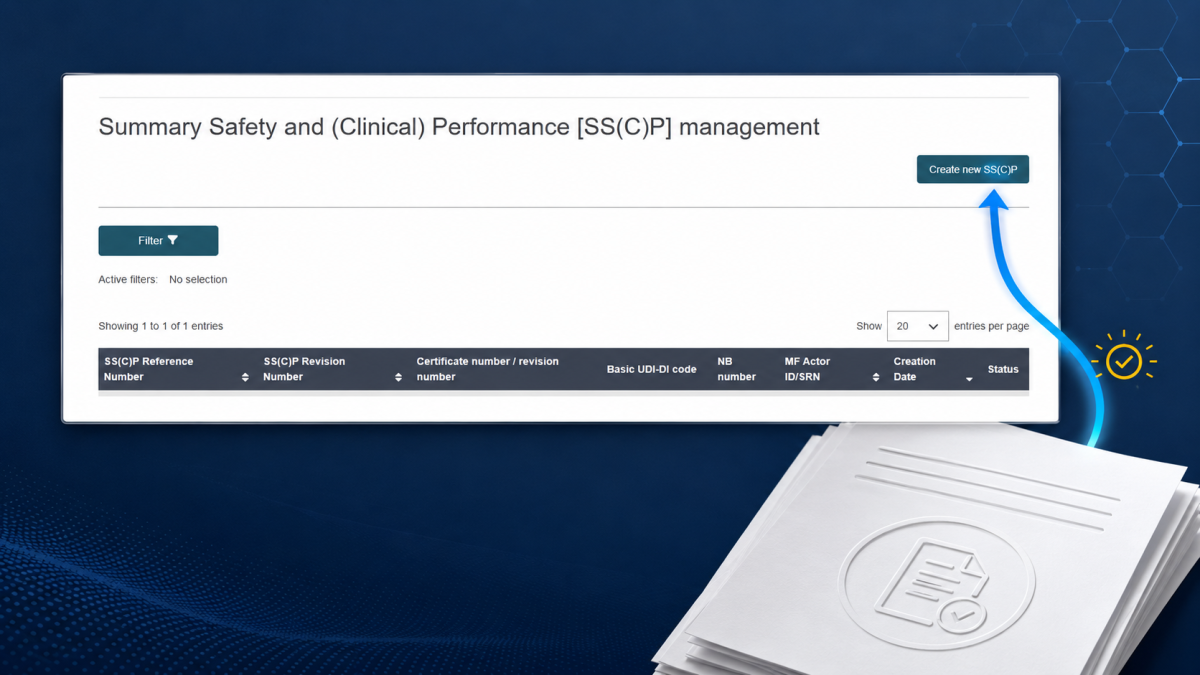
Pregunta: ¿Cómo se registra un solo producto de Clase I?
Registrar un producto individual en EUDAMED: así es como funciona:
Iniciar sesión:
https://webgate.ec.europa.eu/eudamed/Abrir módulo UDI/Devices
Seleccionar"Register new UDI/Device"
Pasarpor el asistente paso a paso (5 pasos):
Paso 1: UDI-DI básico, nombre del dispositivo, EMDN, clase de riesgo, propósito
Paso 2: UDI-DI (GTIN), embalaje, esterilidad
Paso 3: Operadores económicos (fabricante, en su caso, representante autorizado/importador)
Paso 4: Disponibilidad en el mercado (fecha «de», fecha «hasta» si procede)
Paso 5: Validar y enviar (Submit)
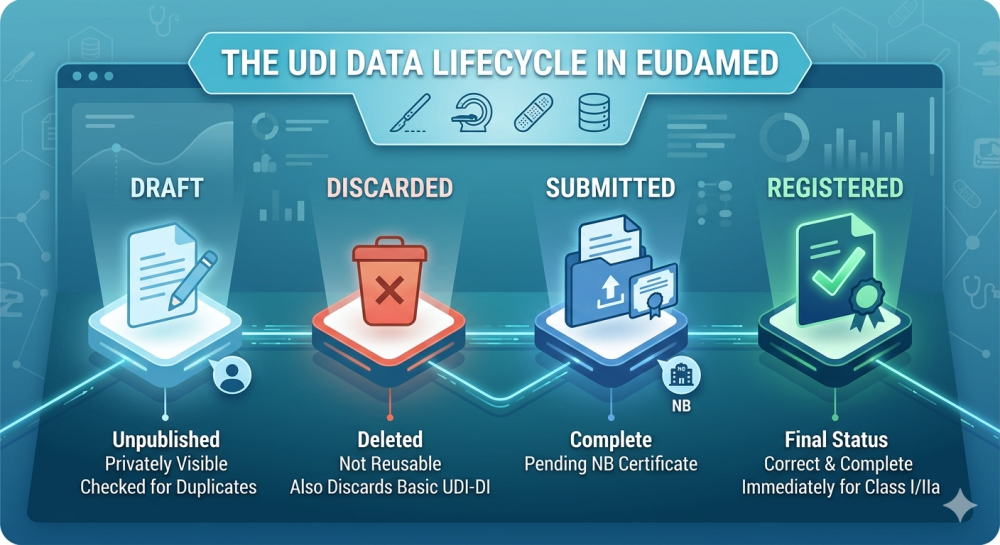
Comprobar estado: el producto se muestra como "Enviado" o "Publicado"
Pregunta: ¿Cómo se registran exactamente los productos en EUDAMED?
Requisito:
Pregunta: ¿Cómo se pueden procesar posteriormente los productos registrados?
✅ Para editar un producto ya registrado en EUDAMED:
Iniciar sesión en EUDAMED
👉 HTTPS://WEBGATE.EC.EUROPA.EU/EUDAMEDAbrir módulo UDI/Devices
Buscar producto a través de UDI-DI o Basic UDI-DI
A la derecha de la entrada, haga clic en → «Acciones» «Actualización»
Recorrer el asistente (Wizard) y modificar los campos deseados
Validar y luego enviar : la actualización se enviará y procesará
📌 Importante:
Solo puedes editar productosvinculados a tu SRN como fabricante
Algunos campos (por ejemplo, Basic UDI-DI) no se pueden cambiar , por lo que es posible que deba crear un nuevo producto
Pregunta: La relación entre las tareas (por ejemplo, cambiar el nombre del producto o añadir un nombre comercial) y las actividades relacionadas con las acciones con la GUI de EUDAMED, así como el orden razonable de las actividades/acciones en la GUI.
Ejemplos típicos de cambios en la GUI
Modificación Campo afectado (GUI) Módulo EUDAMED Acción necesaria Cambiar nombre del producto "Device Name" Módulo UDI/Device Presentación de actualización Añadir nombre comercial "Nombre de la marca" Módulo UDI/Device Presentación de actualización Ajustar número de modelo "Modelo" Módulo UDI/Device Presentación de actualización Ajustar el propósito (Intended Purpose) "Uso previsto" Módulo UDI/Device Presentación de actualización Completar la información de embalaje "Packaging" Módulo UDI/Device Presentación de actualización Modificar datos de esterilidad/etiqueta "Device Characteristics" Módulo UDI/Device Presentación de actualización 🖥️ Proceso en la GUI: secuencia de pasos recomendada
🎯 Escenario: Desea cambiar/añadir el nombre del producto Y el nombre comercial
🔄 1. Iniciar sesión y seleccionar producto
Vaya al módulo UDI/Device
Busque el UDI-DI (GTIN)en cuestión
Haga clic en «Acciones» → «Actualización»
✍️ 2. Ajuste paso a paso en el asistente
La GUI se divide en pasos: los siguientes campos son relevantes:
Paso 1: Información básica de UDI-DI y dispositivos
Aquí puede personalizar Device Name (Nombre del producto) y Brand Name (Nombre comercial)
También puede añadir varios nombres comerciales aquí (por ejemplo, en mercados multilingües)
Paso 2: Características del dispositivo
Compruebe si el cambio de nombre afecta a otros datos (por ejemplo, idioma de la etiqueta, embalaje)
Paso 3–5: Revisión y envío
Después de la última página, puede revisar sus entradas
Haga clic en "Validar" → el sistema comprueba si todos los campos obligatorios son correctos
- A continuación, haga clic en «Enviar»
🔐 3. Después de enviar
- El estado del cambio se mostrará como "Under Review" o "Updated"
- Algunos cambios aparecen inmediatamente, otros solo después de la aprobación de la autoridad competente (según el campo y la implementación nacional)
⚠️ Información importante sobre el orden y la lógica
- Nombre del dispositivo vs. nombre de la marca:
- Nombre del dispositivo = nombre técnico del producto
- Nombre de la marca = marca comercial en el mercado (por ejemplo, "MediPlus®")
- El nombre de la marca puede ser múltiple, el nombre del dispositivo no es el nombre de la → marca, así que elige sabiamente
- Los cambios en el nombre del producto o el nombre de la marca pueden tener efectos retroactivos en la etiqueta, la IFU o la tecnología Documental, es decir, votar internamente de antemano
- Puede agrupar varios cambios en un envío de actualización , esto es más eficienteque enviar todo por separado
Pregunta: ¿Podría dar ejemplos de advertencias críticas o contraindicaciones (paso 4: características UDI-DI)?
En este campo, el fabricante debe indicar advertencias o contraindicaciones relevantes para el producto, si las hubiera. El objetivo es hacer que la seguridad del paciente y del usuario sea directamente visible.
✅ Ejemplos de advertencias críticas típicas:
«No apto para su uso en pacientes con desfibrilador implantado».
"No utilizar cerca de tomógrafos de resonancia magnética (MRI)."
"Utilícelo únicamente por personal especializado capacitado".
«Estéril: no lo utilice si el embalaje está dañado».
✅ Ejemplos de contraindicaciones:
«No usar en caso de alergia conocida al látex».
"No apto para niños menores de 3 años."
«No utilizar en heridas abiertas o enfermedades de la piel en el área de aplicación».
«No utilizar en mujeres embarazadas o en periodo de lactancia».
⚠️ Importante:
Si no hay advertencias o contraindicaciones, el campo se puede rellenar con "Ninguna conocida" , pero nunca se puede dejar en blanco.
La entrada se realiza como texto libre, por lo que debes formularla de forma precisa pero concisa.
Pregunta: Me gustaría saber cuál es la mejor manera de corregir o cambiar un UDI-DI básico en EUDAMED. He intentado crear una nueva entrada con otro Basic UDI-DI para un producto previamente registrado. Sin embargo, recibo un mensaje de error (Paso 3: Información de identificación UDI-DI) porque el código UDI-DI/GTIN del dispositivo ya se ha utilizado para mi entrada anterior.
El UDI-DI básico es la característica de identificación general de un grupo de productos (por ejemplo, la misma finalidad, la clase de riesgo, el diseño esencial).
Está firmemente vinculada al UDI-DI del producto (GTIN) y no se puede intercambiar fácilmente después de la presentaciónsi ya existe una entrada con este GTIN.🔒 ¿Por qué aparece el mensaje de error?
Si intentas registrar un producto con un nuevo Basic UDI-DI pero utilizas un UDI-DI (GTIN) ya presentado con otro Basic UDI-DI, EUDAMED lo detecta como inconsistente y bloquea el proceso:
Error: El código UDI-DI ya se ha utilizado para otro UDI-DI básico.
Opciones de solución:
1. Actualizarentrada existente, no volver a enviar
Si el UDI-DI/GTIN ya está registrado, solo puede ir a través de un cambio en la entrada existente , por ejemplo, a través de un envío de actualización (a través de la interfaz web o M2M/XML), no a través de un nuevo registro.
👉 Pero tenga cuidado: el UDI-DI básico en sí no se puede cambiar con un dispositivo existente.
2. Si el UDI-DI básico era realmente incorrecto (por ejemplo, error tipográfico)
Entonces solo hay dos opciones:
Cerrar sesión en el dispositivo (Deactivate/Delete) y volver a registrarlo con el UDI-DI básico y el UDI-DI correctos
O (si el dispositivo aún no se ha comercializado activamente): generar un nuevo UDI-DI (nuevo GTIN) y volver a registrarlo con el UDI-DI básico correcto
📌 Importante: La conexión UDI-DI ↔ Basic UDI-DI es permanente. No puede existir el mismo GTIN con dos UDI-DIs básicos diferentes .
3. Si se van a registrar varios productos similares:
Luego, cada uno debe recibir su propio GTIN, incluso si difieren en los detalles, y registrarse a través de sus propios UDI-DI básicos, si está justificado (diferente propósito, clase de riesgo, etc.).
Pregunta: ¿Podría describir qué información correcta se debe introducir en el campo ‘Modelo de dispositivo’ (Paso 1: Información básica de UDI-DI)? He notado que los fabricantes no utilizan este campo de manera uniforme en EUDAMED.
Definición según MDCG 2018-1:
El modelo de dispositivo ("Modelo de dispositivo") describe el nombre técnico o comercial del producto bajo el cual se comercializa o utiliza el dispositivo médico de forma identificable. Ayuda a distinguir los productos dentro de un grupo Basic UDI-DI.Por lo tanto, lo✅ correcto sería:
Una designación técnica del modeloque se menciona en la etiqueta, en las instrucciones de uso o en la documentación del producto.
O una designación relacionada con el número de artículo, si no hay un número de modelo disponible.
❌ No son adecuados:
Descripciones del producto («Set de catéteres estériles desechables para niños»)
Finalidades ("Para el tratamiento de XYZ")
Textos libres con palabras clave o combinaciones de variantes
🔍 Ejemplos de entradas de modelo de dispositivo correctas:
Tipo de producto Modelo de dispositivo Marcapasos Modelo ACURA 3000 Mascarilla quirúrgica desechable Tipo 2R – MaskCare Blue Implantes dentales IMPL-24-Titanium Software (SaMD) Diagware v1.4.2 Tensiómetro BP-Monitor Pro M120
Pregunta: ¿Qué elementos de datos son obligatorios para el registro del producto?
Campos obligatorios en el registro del producto en EUDAMED (nivel UDI-DI)
Aquí ordenados por categorías:📘 A. Información básica del dispositivo (paso 1 de la GUI)
Campo ¿Deber? Comentario Basic UDI-DI ✅ Sí Asignado por el propio fabricante (por ejemplo, a través de GS1) Nombre del dispositivo ✅ Sí Nombre técnico (sin texto de marketing) Nombre de la marca ❌ No Solo si es utilizado por el mercado Código EMDN ✅ Sí Código de nomenclatura de la UE (mín. 4 dígitos) Estado regulador (MDR/IVDR) ✅ Sí Indica si MDR o IVDR Clase de riesgo ✅ Sí I, IIa, IIb, III (MDR) o A–D (IVDR) Propósito previsto ✅ Sí Finalidad prevista en texto sin formato Notified Body Information ✅ Sí, si procede Solo para productos con certificado NB 📦 B. UDI-DI & Packaging (Paso 2 de la GUI)
Campo ¿Deber? Comentario UDI-DI (GTIN o similar) ✅ Sí Identificador principal en el embalaje Tipo de envase primario ✅ Sí Por ejemplo, embalaje individual, a granel, etc. Sterility Information ✅ Si el producto es estéril De lo contrario, puede permanecer vacío Storage & Shelf Life ❌ Opcional Solo en caso de dependencia de la temperatura/caducidad Advertencias críticas/ contraindicaciones ❌ Opcional Si está disponible, introduzca 👥 C. Operadores económicos (paso 3 de la GUI)
Campo ¿Deber? Comentario Fabricante (SRN) ✅ Sí Automático, si tú mismo eres fabricante Representante autorizado ✅ Si no eres fabricante de la UE Debe tener SRN válidos Importador ❌ Opcional Solo si está disponible, de lo contrario, déjelo vacío 🌍 D. Información de mercado (paso 4 de la GUI)
Campo ¿Deber? Comentario Fecha «De» (Date from) ✅ Sí Fecha de inicio de la disponibilidad del mercado Fecha «Hasta» (Date to) ❌ No Solo en caso de baja/retirada del mercado 📎 E. Anexos (paso 5 de la GUI)
Campo ¿Deber? Comentario Subir etiqueta / IFU ❌ Opcional Puede ser requerido en auditorías, pero no es obligatorio Documento técnico ❌ No directamente en EUDAMED Solo se requiere en caso de solicitud de las autoridades
Pregunta: Solicito aclaraciones con respecto a la sección “Información de mercado” en EUDAMED, en particular con respecto a los campos de fecha “desde (AAAA-MM-DD)” y “hasta (AAAA-MM-DD)”. ¿Qué significan exactamente estos datos o deben completarse?
Depende de la legislación bajo la que registre su producto. Si selecciona la legislación antigua (MDD, IVDD, AIMDD), debe introducir al menos la fecha "válida hasta" (opcionalmente, también puede especificar la fecha de inicio). Pero solo la fecha "hasta" es obligatoria. Por el contrario, si selecciona el nuevo reglamento como «MDR» o «IVDR», no tendrá que indicar ninguna fecha.
¿Qué significan los campos "desde (YYYY-MM-DD)" y "hasta (YYYY-MM-DD)" en la sección "Información de mercado"?
Campo Significado Fecha «de» ( Date from which the device is/was made available on the market)La fecha a partir de la cual el producto se puso a disposición por primera vez en un mercado de la UE (de acuerdo con el Artículo 2 No. 27 MDR/IVDR: "Comercialización") Fecha «hasta» ( Date until which the device is/was made available on the market)La fecha en la que el producto está o ha estado activo en el mercado, por ejemplo, en caso de retirada del mercado, salida del producto, desactivación 🧠 ¿Qué significa esto en la práctica?
La fecha «de» suele ser la fecha de lanzamiento en la UE
La fecha «hasta» solo se establececuando el producto ya no se comercializa activamente o tiene una fecha de caducidad fija
📌Ejemplo:un producto se lanzó el 01/01/2023 y todavía está disponible:
→ "de" = 2023-01-01 "
a→" = dejar en blanco (no es necesario)⚠️ ¿Los campos son obligatorios?
▶️ Estado actual según la Comisión de la UE (XSD v2.0 y especificación GUI):
Campo ¿Campo obligatorio? Fecha «de» ✅ Sí, obligatorio Fecha «hasta» ❌ No, solo se requiere en caso de descontinuación o finalización del mercado
Pregunta: ¿ La sección 2 "Lista de todos los países en los que el producto está o estará disponible" se aplica solo a los países de la UE?
Sí, en esta sección solo se pueden seleccionar los países de la UE.
Pregunta: Indicación de que un país debe definirse como "originalmente comercializado", ¿qué se entiende por damir? ¿El país en el que el dispositivo médico (conjunto de datos UDI) se introdujo por primera vez en el mercado?
¿Esta información se refiere exclusivamente a un primer país miembro de la UE o también pueden tenerse en cuenta en este campo terceros países no pertenecientes a la UE?
EUDAMED se refiere exclusivamente al mercado de la UE. Los países fuera de la UE no forman parte de este sistema. Los fabricantes que deseen distribuir sus productos también en países no pertenecientes a la UE deben cumplir los requisitos reglamentarios respectivos de estos países por separado.
Pregunta: ¿Es aconsejable que los productos se combinen y registren de acuerdo con los UDI básicos? (agrupados o por separado), por ejemplo, mangueras para insufladores – Palabra clave: ¿Es posible agrupar productos para minimizar la carga de trabajo?
Sí, no solo se permite una agrupación razonable («agrupación») de productos bajo un UDI-DI básico común, sino que también se prevé expresamente, siempre que se cumplan ciertos criterios. Esto reduce significativamente el esfuerzo de registro y mantenimiento en EUDAMED.
📘 ¿Qué es el Basic UDI-DI y para qué sirve?
El UDI-DI básico es:
la clave principal para el grupo de productos
el enlace a la declaración de conformidad, certificación, técnica Documentación
no se imprime en el embalaje , sino solo en EUDAMED y Doku
📦 ¿Cuándo se pueden agrupar los productos bajo un UDI-DI básico común?
Según el MDCG 2018-1 (y la Comisión Europea):
✅ Los productos deben:
Criterio Significado Misma finalidad Cumplen la misma finalidad clínica o diagnóstica Misma clase de riesgo por ejemplo, toda la clase IIa Construcción/diseño básico similar por ejemplo, mangueras del mismo material, misma conexión Parte de la misma tecn. Documentación Se evalúan en un archivo, por ejemplo, juntos en un DoC 🧪 Ejemplo: mangueras para insufladores
Estos se pueden agrupar fácilmente si:
tienen la misma función (por ejemplo, conexión del insuflador con el paciente)
solo difieren en longitud, color o unidad de embalaje
el mismo fabricante los produce y existe un documento técnico común
👉 Entonces es aconsejable agrupar estas variantes bajo un UDI-DI básico y registrar varios UDI-DI (GTIN) bajo esta base.
🎯 Ventajas del clúster (agrupación):
Ventaja Descripción 🔁 Menos esfuerzo Solo un registro a nivel de base con varios UDI-DI 🧾 Documento unificado Solo se requiere una declaración de conformidad y un expediente técnico 🔍 Mejor visión general En EUDAMED puedes gestionar tus variantes de forma más estructurada 📈 Escalabilidad Las nuevas variantes se pueden añadir más fácilmente más adelante ⚠️ Pero tenga cuidado: cuando no debe agruparse:
No agrupar en… Motivo Diferentes finalidades por ejemplo, manguera para CO₂ vs. para otros gases Diferentes clases de riesgo p. ej., clase I frente a clase IIa Diferentes evaluaciones regulatorias por ejemplo, una variante es estéril y la otra no Diferentes certificados o cuerpos notificados se requieren dipositivos UDI básicos→ separados
Pregunta: ¿Podemos utilizar el disco UDI básico de GS1 para dispositivos heredados o tenemos que dejar que Eudamed lo genere? ¿Esto significa que estos últimos deben estar vinculados a los nuevos?
Sí, también puede utilizar los UDI-DI básicos generados por GS1 para dispositivos heredados; no es necesario que EUDAMED los genere automáticamente.
👉 Debe asignar el UDI-DI básico usted mismo (por ejemplo, a través de GS1), también para productos heredados , si desea registrarlo en EUDAMED (voluntario u obligatorio más adelante).
Pregunta: ¿Existe la posibilidad de generar nuevos códigos EMDN si un nuevo producto con una nueva tecnología no se puede encontrar en los códigos existentes?
No, los fabricantes no pueden generar sus propios códigos EMDN. Los nuevos códigos solo pueden ser añadidos por la Comisión de la UE, previa solicitud o a través de canales definidos.
¿Qué hacer si no hay un código EMDN adecuado?Seleccionar el código EMDN más cercano
Está obligado a elegir el código existente más adecuado
Por ejemplo, si su producto es una innovadora máquina de diálisis portátil, pero solo hay códigos para máquinas de diálisis clásicas, tome la más cercana
Por lo general, los códigos de 4 o 6 dígitos (nivel 2 o 3) son suficientes para el registro del producto
📌 Esta selección debe estar bien documentada, incluso para las auditorías (por ejemplo, por qué has elegido este código).
Pregunta: ¿Qué más se puede hacer con la GUI, por ejemplo, puede buscar productos específicos, clases de productos, fabricantes, importadores, etc.? La búsqueda a menudo da respuestas vacías.
Funciones típicas en el módulo UDI/Device:
Función Descripción Comentario 🔍 Buscar productos Según UDI-DI (GTIN), Basic UDI-DI, Device Name, Brand Name A menudo sensibles a los casos y muy restrictivas 🗂️ Ver detalles del producto Todos los detalles del producto registrado incl. Embalaje, etiqueta, uso previsto, etc. Solo si el resultado de la búsqueda es correcto ✏️ Cambiar producto (Update Submission) Modificar o completar la información del producto Solo para productos procedentes de su propio SRN 🗑️ Desactivar producto (Deactivate) Retirar o retirar el producto del mercado Solo es posible por el fabricante registrado ⬆️ Registrar nuevo producto Con asistente completo de 5 pasos Basic UDI-DI debe ser correcto Ⅰ Comprobar el estado del producto "Submitted", "Under Review", "Published", etc. Resumen importante del estado del flujo de trabajo Funciones típicas en el módulo Actor:
Función Descripción Comentario 🔍 Buscar actores Por SRN, nombre de la empresa, función (fabricante, importador, etc.) A menudo solo funciona con la ortografía exacta 🔗 Vinculación con otros actores p. ej., fabricante con importador o representante autorizado Se requiere confirmación mutua 🧾 Mostrar detalles del actor Dirección, roles, responsabilidades, etc. Útil para las autoridades o pruebas de la cadena de suministro ⚠️ ¿Por qué la búsqueda a menudo no da resultados?
Estas son las causas y consejos más comunes:
1. Escritura incorrecta/ falta de formato
- EUDAMED distingue entre mayúsculas y minúsculas
🔍Acura3000yacura30002. Los términos parciales no funcionan
- No hay búsqueda automática de "Contiene"→, por ejemplo,
Medino encuentraMediPlus 5000
👉 Si es posible, utiliza el nombre completo3. SRN introducido incorrectamente
- Los SRN deben introducirse en el formato exacto (por ejemplo,
DE-MF-000000xxx)4. Sin acceso a entradas ajenas
- Solo puedes ver los productos/actoresvinculados a tu organizaciónLos
datosvisibles👉 públicamente solo llegan después de la conexión completa en vivo de EUDAMED5. Sesiones obsoletas/caducadas
- La GUI tiene un tiempo de esperadespués del cual ya no se muestran los resultados reales; a menudo ayuda: volver a iniciar sesión
Consejos para una búsqueda exitosa en la GUI:
Lo que buscas Como introducirlo UDI-DI / GTIN Introduzca el número completo exactamente Basic UDI-DI Sin espacios ni caracteres especiales Nombre / modelo del dispositivo Exactamente como en la entrada EUDAMED Fabricante / Importador (SRN) Completo con código de país (DE-MF-...)Estado Opcional: búsqueda solo de productos activos ( publicados)
Pregunta: ¿Hay una subida a través de Excel? ¿Cómo podemos evitar el doble mantenimiento de datos, en Eudamed e internamente?
❌ **No: actualmente EUDAMED no admite la carga directa de Excel.
La Comisión de la UE solo permite:
Entrada manual a través del GUI (portal web)
Carga automatizada a través de archivo XML de acuerdo con el esquema XSD oficial (para registro masivo o M2M)
💡 Pero: puede trabajar con la plantilla de Excel de Europe IT
¿Cómo evitar el doble mantenimiento de datos?
Aquí también podemos recomendar nuestra plantilla Excel.
Pregunta: ¿Recomienda herramientas específicas para el registro de productos en los que tenemos el rol de fabricante?
Sí, recomendamos la solución interna de Europe IT Consulting, basada en una plantilla de Excel estructurada.
No dude en ponerse en contacto con nosotros.
Pregunta: ¿Reemplazará Eudamed el registro en bases de datos locales una vez que los módulos estén en pleno funcionamiento o no?
Probablemente sí. Suponemos que una vez que todos los módulos EUDAMED estén completamente operativos y se hayan publicado oficialmente en el Diario Oficial de la UE (OJEU), EUDAMED reemplazará las bases de datos nacionales de productos para productos compatibles con MDR/IVDR.
Sin embargo, durante la fase de transición, muchos Estados miembros mantienen sus sistemas locales en paralelo.¿Qué significa esto exactamente?
📌 Una vez que EUDAMED esté completamente operativo (según la publicación de OJEU):
Zona Jurisdicción Registro UDI/Device central en EUDAMED – sustituye a las bases de datos nacionales Registro de actores (SRN, etc.) Solo sobre EUDAMED Vigilancia y vigilancia del mercado Visible en toda la UE a través de EUDAMED Vinculación de fabricantes, importadores, apoderados Solo válido en EUDAMED 📅 Pero hasta entonces, se aplica lo siguiente: Fase de transición = estructuras paralelas nacionales
Muchas autoridades (por ejemplo, BfArM en DE, FAMHP en BE) continúan operando sus propias bases de datos:
Ejemplo Estado actual Alemania (BfArM) Sigue siendo necesario el registro nacional de equipos usados y ciertos IVDs Francia (ANSM) Requiere en parte entradas nacionales adicionales, por ejemplo, sobre la disponibilidad del mercado Italia (Ministerio) Transferencia de datos fija planificada de EUDAMED al sistema nacional 📌 ¿Qué significa esto para vosotros en la práctica?
Fase Qué hacer Ahora (2025, uso voluntario de EUDAMED) Utilizar EUDAMED = ventaja estratégica (preparación, seguridad de la auditoría) A partir del anuncio OJEU (previsto para 2025) 24 meses para la migración completa Después (uso obligatorio) Solo queda el registro en EUDAMED: los sistemas nacionales pueden servir solo para fines de supervisión o complementarios
Pregunta: ¿Qué bases de datos locales deben mantenerse (por ejemplo, el fabricante Ort – Alemania – BfArM/DMIDS) y otros?
Aunque EUDAMED se convertirá en el sistema central a medio plazo, actualmente (2025) existen varias bases de datos nacionales en la UE que deben mantenerse, dependiendo del tipo de producto, la ubicación y los requisitos del mercado.
Aquí hay una lista clara de las bases de datos nacionales más importantes que aún deben mantenerse activamente (a partir de 2025), especialmente para los fabricantes con sede en la UE:🇩🇪 España – BfArM / DMIDS
Base de datos Finalidad ¿Deber? DMIDS (Sistema Alemán de Información y Base de Datos de Dispositivos Médicos) Registro de fabricantes, apoderados, empaquetadores de sistemas/procedimientos ✅ Sí, sigue siendo obligatorio, hasta que EUDAMED sea oficialmente obligatorio Mensaje DIMDI para equipos antiguos (MDD/IVDD) Notificación de dispositivos heredados ✅ Sí, si todavía hay productos MDD/IVDD en el mercado Mensaje UDI ❌ Será sustituido en perspectiva por EUDAMED 📌 Los fabricantes con sede en DE actualmente tienen que mantener EUDAMED y DMIDS en paralelo, especialmente para equipos antiguos o vigilancia del mercado nacional.
🇫🇷 Francia – ANSM
Base de datos Finalidad ¿Deber? Base de données des dispositifs médicaux (BDDM) Registro nacional de productos, principalmente IVDs ✅ Sí Formulario ANSM de disponibilidad de mercado Información a las autoridades sobre la comercialización en Francia ✅ Sí, también en el registro EUDAMED 🇮🇹 Italia – Ministero della Salute
Base de datos Finalidad ¿Deber? Repertorio Dispositivi Medici (RDM) Registro obligatorio para la venta en Italia ✅ Sí, incluso con el registro EUDAMED adicionalmente Notificación mediante códigos CND Italia utiliza CND en lugar de EMDN – se debe convertir ✅ Sí 🇪🇸 España – AEMPS
Base de datos Finalidad ¿Deber? Registro de Productos Sanitarios (RPS) Obligación de registro antes de la comercialización en España ✅ Sí 🇧🇪 Bélgica – FAMHP / AFMPS
Base de datos Finalidad ¿Deber? Portal web de registro de actores Registro del fabricante, incluso durante el uso de EUDAMED ✅ Sí Mensaje del producto Sigue siendo necesario para la vigilancia del mercado nacional ✅ Sí, temporal 🇦🇹 Austria – BASG
Base de datos Finalidad ¿Deber? Portal MedProd de BASG Notificación de productos, especialmente en caso de distribución por parte de organismos austriacos ✅ Sí, paralelamente a EUDAMED 🔁 ¿Qué significa esto en la práctica?
Si… Entonces tienes que… Su domicilio social se encuentra en Alemania Entrada DMIDS + dispositivos antiguos en DIMDI si es necesario Distribuyen productos en Francia, Italia, España, Bélgica, Austria Realizar registro nacional de producto además de EUDAMED Solo venden en la zona del tejado Mantener solo BfArM y BASG, hasta que EUDAMED se active