
Master UDI-DI in EUDAMED:
welche Produkte Hersteller jetzt registrieren müssen – und welche noch nicht
Executive Summary
- Kontaktlinsen ab 9. November 2026,
- Brillenprodukte ab 1. November 2028.
Diese Sonderregel verschiebt aber nicht die Registrierungspflicht für alle anderen Produkte.
1. Warum herrscht aktuell Unsicherheit?
Die Unsicherheit entsteht aus zwei parallelen Entwicklungen: einerseits wurde EUDAMED für die ersten vier Module verbindlich. Andererseits ist die technische und regulatorische Master-UDI-Logik für bestimmte hoch individualisierte Produkte zeitlich versetzt eingeführt worden.
Viele Hersteller fragen deshalb zurecht: muss ich Produkte jetzt in EUDAMED registrieren, obwohl die Master UDI-DI für meine Produktgruppe erst später kommt? Die fachlich richtige Antwort lautet: es kommt auf den Produkttyp an.
Kernaussage: Die Master UDI-DI ist keine allgemeine Verschiebung der EUDAMED-Pflichten. Sie ist eine Sonderlogik für bestimmte hoch individualisierte MDR-Produkte. Normale MDR-/IVDR-Produkte bleiben von dieser Sonderlogik unberührt.
2. Allgemeine EUDAMED-Pflicht seit 28. Mai 2026
Die Europäische Kommission bestätigt, dass seit dem 28. Mai 2026 vier EUDAMED-Module verpflichtend zu nutzen sind: Actor Registration, UDI/Device Registration, Notified Bodies & Certificates und Market Surveillance. Die Kommission verweist dabei auf den Beschluss (EU) 2025/2371, der am 27. November 2025 im Amtsblatt der Europäischen Union veröffentlicht wurde und den sechsmonatigen Übergangszeitraum ausgelöst hat.
Das UDI/Device-Modul ist damit für die Produktregistrierung zentral. Hersteller müssen die UDI-/Device-Informationen der Produkte übermitteln, die sie auf dem EU-Markt in Verkehr bringen. Für Produkte unter MDR und IVDR erfolgt die Registrierung im Grundsatz auf Ebene des Device Identifier, also insbesondere UDI-DI; Produktionskennzeichen wie Chargennummer oder Seriennummer werden nicht als eigene Produktregistrierung pro Einzelstück registriert.
Neue Produkte ab dem 28. Mai 2026
Wird die erste verkaufsfähige Einheit eines Regulation Device oder Systems/Procedure Pack mit einer bestimmten UDI-DI am oder nach dem 28. Mai 2026 auf dem EU-Markt in Verkehr gebracht, muss die Registrierung im UDI/Device-Modul vor diesem ersten Inverkehrbringen erfolgt sein.
Produkte, die bereits vor dem 28. Mai 2026 in Verkehr gebracht wurden
Wurde die erste Einheit eines Legacy Device oder Regulation Device bereits vor dem 28. Mai 2026 in Verkehr gebracht und werden weitere Einheiten derselben UDI-DI am oder nach dem Pflichtdatum weiterhin in Verkehr gebracht, gilt eine Nachregistrierungsfrist. Nach der EU-Übergangsübersicht endet diese Frist am 28. November 2026.
Praxisregel: Für normale MDR-/IVDR-Produkte sollte kein Hersteller auf die Master UDI-DI warten. Die Pflicht zur UDI-/Device-Registrierung ist bereits aktiv.
3. Was ist die Master UDI-DI?
Die Master UDI-DI wurde für Produkte mit einem hohen Individualisierungsgrad eingeführt. Bei solchen Produkten könnten sehr viele Varianten entstehen, obwohl sie regulatorisch und technisch stark vergleichbar sind. Eine normale UDI-DI je Variante würde zu einer sehr großen Anzahl an Datensätzen führen.
Die Master UDI-DI soll vergleichbare Produktvarianten anhand definierter Parameter unter einem gemeinsamen Identifier zusammenfassen. Sie wirkt bei den betroffenen Produktgruppen funktional wie eine besondere Form des Device Identifier für EUDAMED.
Aktuell betrifft die Master UDI-DI insbesondere folgende MDR-Produktgruppen:
- Standard soft contact lenses
- Standard Rigid Gas Permeable contact lenses
- Made to order soft contact lenses
- Made to order Rigid Gas Permeable contact lenses
- Spectacle frames
- Spectacle lenses
- Ready-made reading spectacles
Wichtig: Die Master UDI-DI betrifft nach aktuellem Stand MDR-Produkte. Für IVDR-Produkte ist auf den offiziellen Kommissionsseiten keine vergleichbare Master-UDI-Sonderlogik beschrieben. IVD-Hersteller sollten daher grundsätzlich von der normalen UDI-/Device-Registrierungslogik nach IVDR ausgehen.
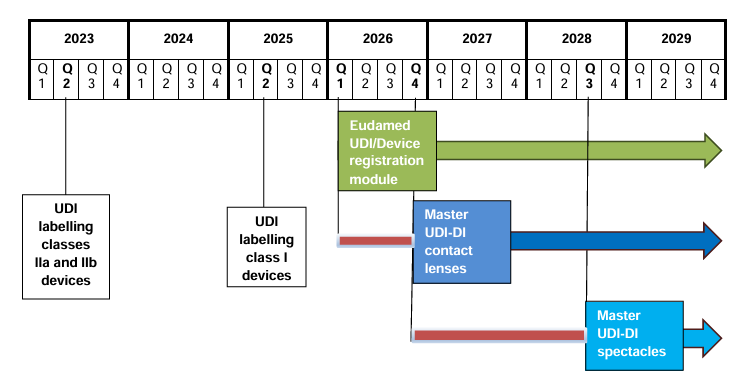
4. Welche Fristen gelten?
| Produkt- / Fallgruppe | Registrierung in EUDAMED | Maßgeblicher Zeitpunkt | Praktische Bewertung |
|---|---|---|---|
| Normale MDR-/IVDR-Produkte erstes Inverkehrbringen ab 28.05.2026 |
Pflicht | Vor dem ersten Inverkehrbringen | UDI-/Device-Registrierung muss vor Markteintritt abgeschlossen sein. |
| Legacy- oder Regulation Devices erste Einheit vor 28.05.2026, weitere Einheiten danach |
Pflicht | Bis spätestens 28.11.2026 | Backlog systematisch prüfen und rechtzeitig nachregistrieren. |
| Kontaktlinsen | Sonderlogik | Master UDI-DI ab 09.11.2026 | Registrierung folgt der Master-UDI-Pflicht. Vorbereitung jetzt erforderlich; freiwillige frühere Zuweisung ist möglich. |
| Brillenfassungen, Brillengläser, Fertiglesebrillen | Sonderlogik | Master UDI-DI ab 01.11.2028 | Derzeit keine normale UDI-DI-Registrierung erzwingen. EUDAMED blockiert diese Special Device Types aktuell systemseitig. |
| Produkte, die nach dem Pflichtdatum nicht mehr in Verkehr gebracht werden | Grundsätzlich keine Registrierung | Nur relevant bei PMS-/Vigilanz-Aktion | Keine Registrierung im UDI/Device-Modul erforderlich, sofern keine PMSV-Aktion ausgelöst wird. |
| Custom-made devices | Nicht im UDI/Device-Modul | Nur begrenzter Datensatz bei Vigilanzbedarf | Nicht als normale UDI-/Device-Registrierung behandeln. |
5. Kontaktlinsen: Master UDI-DI ab 9. November 2026
Für Kontaktlinsen wurde die Master UDI-DI bereits durch die delegierte Verordnung (EU) 2023/2197 eingeführt; die Anwendung wurde durch die delegierte Verordnung (EU) 2025/788 auf den 9. November 2026 verschoben. Das MDCG-Positionspapier 2025-7 Rev. 1 bestätigt, dass die Master-UDI-Zuweisung für Kontaktlinsen ab diesem Datum umzusetzen ist.
Entscheidend ist die MDCG-Position zur Interaktion mit EUDAMED: Für die betroffenen hoch individualisierten Produkte folgt die Pflicht zur Kennzeichnung mit Master UDI-DI und zur Registrierung im UDI/Device-Modul der jeweiligen Master-UDI-Zuweisungspflicht. Kontaktlinsen, die vor dem 9. November 2026 produziert wurden, müssen nach dieser Position nicht zwingend eine Master UDI-DI auf dem Label tragen.
Gleichzeitig wird Herstellern empfohlen, die freiwillige frühere Zuweisung der Master UDI-DI zu nutzen, sobald sie praktisch möglich ist. Wird eine Master UDI-DI freiwillig zugewiesen, sollen Labeling und EUDAMED-Registrierung entsprechend folgen.
Empfehlung für Kontaktlinsenhersteller: nicht abwarten, bis der Stichtag erreicht ist. Jetzt sollten Gruppierungslogik, Parameterbereiche, UDI-Ausgabestelle, Labeling-Auswirkungen, EUDAMED-Datenmodell und QMS-Dokumentation vorbereitet werden.
6. Brillenprodukte: Master UDI-DI ab 1. November 2028
Für Brillenfassungen, Brillengläser und Fertiglesebrillen gilt nach der delegierten Verordnung (EU) 2025/1920 eine spätere Umsetzung. Die Master-UDI-Zuweisung soll für diese Produktgruppen ab dem 1. November 2028 verpflichtend werden.
Zusätzlich gibt es eine wichtige technische Besonderheit: Das EUDAMED Information Centre weist darauf hin, dass bestimmte Special Device Types derzeit nicht registriert werden können. Werden Spectacle frames, Spectacle lenses oder Ready-made reading spectacles ausgewählt, blockiert das System die Speicherung. Eine Registrierung per Machine-to-Machine oder Bulk Upload ist ebenfalls nicht möglich.
Empfehlung für Hersteller von Brillenprodukten: keine Ersatzregistrierung mit falscher Produktlogik versuchen. Stattdessen Master-UDI-Datenmodell, Produktparameter, ERP-/PLM-Struktur, Basic-UDI-Gruppierung und QMS-Traceability vorbereiten und die Systemfreigabe durch die Kommission beobachten.
7. Zeitleiste
- 27. November 2025:
Veröffentlichung des Beschlusses (EU) 2025/2371 im Amtsblatt. Damit wird die Funktionsfähigkeit der ersten vier EUDAMED-Module festgestellt und der sechsmonatige Übergangszeitraum ausgelöst.
- 28. Mai 2026:
Actor Registration, UDI/Device Registration, Notified Bodies & Certificates und Market Surveillance werden verpflichtend.
- 9. November 2026:
Master UDI-DI für Kontaktlinsen wird verpflichtend.
- 28. November 2026:
Deadline zur Registrierung bestimmter Legacy- und Regulation Devices, die bereits vor dem Pflichtdatum in Verkehr gebracht wurden und danach weiter vermarktet werden.
- 1. November 2028:
Master UDI-DI für Brillenfassungen, Brillengläser und Fertiglesebrillen wird verpflichtend.
8. Was Hersteller jetzt konkret tun sollten
1. Portfolio sauber segmentieren
Die wichtigste Sofortmaßnahme ist eine klare Trennung des Portfolios. Normale MDR-/IVDR-Produkte, Kontaktlinsen, Brillenprodukte, Legacy Devices, Custom-made Devices und nicht mehr in Verkehr gebrachte Produkte dürfen nicht nach derselben Logik behandelt werden.
2. UDI-/Device-Backlog priorisieren
Für normale MDR-/IVDR-Produkte sollte der UDI-/Device-Backlog sofort geprüft werden. Besonders relevant sind Basic UDI-DI, UDI-DI, EMDN-Code, Risikoklasse, Handelsname, Modell-/Katalognummern, Zertifikatsbezug, Herstellerdaten und gegebenenfalls Authorized Representative oder Importeur.
3. Master-UDI-Datenmodell vorbereiten
Hersteller von Kontaktlinsen und Brillenprodukten sollten die Master-UDI-Logik nicht erst kurz vor dem Stichtag aufbauen. Notwendig sind klare Gruppierungsregeln, definierte Parameterbereiche, Abstimmung mit der UDI-Ausgabestelle, Anpassungen in ERP/PLM sowie interne Verantwortlichkeiten für Datenfreigabe und Datenqualität.
4. Keine falschen Ersatzregistrierungen erzeugen
Bei Brillenprodukten ist besondere Vorsicht geboten. Wenn EUDAMED bestimmte Special Device Types systemseitig blockiert, sollten Hersteller nicht versuchen, über unpassende Ersatzkategorien eine Registrierung zu erzwingen. Das kann später zu Dateninkonsistenzen und Korrekturaufwand führen.
5. QMS- und Audit-Readiness sicherstellen
EUDAMED-Daten sind nicht nur IT-Daten. Sie müssen mit technischer Dokumentation, Declaration of Conformity, Labeling, Zertifikaten und internen Stammdaten übereinstimmen. Abweichungen zwischen regulatorischer Dokumentation und EUDAMED-Datensatz können in Audits und Zertifizierungsprozessen problematisch werden.
9. Fazit
Die Situation ist differenziert, aber nicht beliebig. Seit dem 28. Mai 2026 gilt die EUDAMED-Pflicht für die ersten vier Module. Hersteller normaler MDR-/IVDR-Produkte müssen ihre Produkte grundsätzlich jetzt registrieren: neue Produkte vor dem ersten Inverkehrbringen, bestimmte bereits vor dem Pflichtdatum in Verkehr gebrachte Produkte bis spätestens 28. November 2026.
Die Master UDI-DI verschiebt diese allgemeine Pflicht nicht. Sie schafft aber eine Sonderlogik für bestimmte hoch individualisierte MDR-Produkte. Kontaktlinsen folgen der Master-UDI-Pflicht ab 9. November 2026. Brillenfassungen, Brillengläser und Fertiglesebrillen folgen ab 1. November 2028; zudem ist deren Registrierung in EUDAMED derzeit systemseitig blockiert.
Hersteller sollten daher nicht pauschal fragen, ob EUDAMED „schon gilt“ oder „noch nicht gilt“. Die richtige Frage lautet: Welche Produktgruppe liegt vor, welche UDI-Logik gilt und welcher Stichtag ist für genau diese Produktgruppe maßgeblich?
Quellen und weiterführende Dokumente
- Europäische Kommission: EUDAMED Overview
- Europäische Kommission: EUDAMED UDI/Device Registration
- Europäische Kommission / MDCG: Q&A on the gradual roll-out of EUDAMED pursuant to Regulation (EU) 2024/1860
- Europäische Kommission: EUDAMED registration of devices – transition period
- MDCG 2025-7 Rev. 1: Timelines of the implementation of Master UDI-DI to contact lenses and spectacle products
- EUDAMED Information Centre: Basic UDI-DI information – Special Device Types
- EUR-Lex: Commission Decision (EU) 2025/2371
- EUR-Lex: Regulation (EU) 2024/1860
Dieser Fachbeitrag basiert auf öffentlich verfügbaren regulatorischen Informationen mit Stand 17. Juni 2026. MDCG-Dokumente und Q&A-Dokumente dienen der praktischen Auslegung und Orientierung; verbindliche Auslegung des Unionsrechts bleibt den zuständigen Gerichten vorbehalten. Der Beitrag ersetzt keine individuelle Rechts- oder Regulierungsberatung.






Related Posts