
Schlüsselwörter: MDR, IVDR, EUDAMED, Module, ACT, UDI, CRF, CIPS, VGL, MSU
Was ist die EUDAMED?
Die EUDAMED steht für „European Database for Medical Devices“ wird von der Europäischen Kommission betrieben und dient dazu alle relevanten Informationen zu Medizinprodukten auf dem EU Markt zu zentralisieren sowie die Rückverfolgbarkeit und Transparenz zu gewährleisten.
Die EUDAMED geht auf einen Beschluss der EU-Kommission (2010/227/EU) zurück. In diesem Beschluss formuliert die EU den Zweck der EUDAMED:
Am Beispiel des PIP-Skandals (Verbot von Brustimplantaten eines franz. Herstellers) kann man erkennen, dass eine zentrale Verwaltung von Daten, wie klinische Studien, Marktüberwachung und Rückrufe, durchaus Sinn ergeben und Leben retten können.
- Den genauen Zeitplan der MDR/IVDR finden Sie hier
- UDI Fachbegriffe Index finden Sie hier
- UDI Lösung für FDA / US / GUDID finden Sie hier
Was wird in der EUDAMED gespeichert?
Die EUDAMED wird insgesamt 6 vernetzte Bereiche umfassen, die alle elektronisch zugänglich für die Medizinprodukte Hersteller sein werden. Somit können alle Informationen voll elektronisch übertragen werden.
Die Europäische Kommission, die zuständigen Behörden und die Mitgliedstaaten können alle Modulinformationen einsehen/darauf zugreifen.
Die 6 Module der EUDAMED bestehen aus:

EUDAMED 3 Database
 1. Modul ACT – Actor Registration
1. Modul ACT – Actor Registration
Dieses Modul ermöglicht, dass sich Hersteller (Akteure) registrieren und später eindeutig identifiziert werden können. Zu den Akteuren werden Hersteller, Importeuere und EU Repräsentanten gezählt. Nach der Registrierung und Überprüfung der angegebenen Daten erhalten die Akteure einen Zugang zur EUDAMED. Mit Hilfe der SRN (Single Registration Number) kann jeder Wirtschaftsakteur eindeutig identifiziert werden. Die SRN ist allerdings getrennt bei einer Benannten Stelle (CA) zu beantragen. Die Benannten Stellen müssen sich von der EU akkreditieren lassen. Die Liste aller zugelassenen Benannten Stellen kann man hier nachschauen.
Öffentlicher Zugang. Seit dem 1. Dezember 2020 sind alle in EUDAMED registrierten Akteursinformationen öffentlich zugänglich, zusammen mit der eingeschränkten EUDAMED-Website.
2. Modul UDI – Unique Device Identification
Das UDI Modul wird alle gerätespezifische Informationen enthalten und die gleichen Funktionen wie die vergleichbare Datenbank der amerikanischen Gesundheitsbehörde (FDA) GUDID haben. Der Hauptunterschied zur GUDID besteht darin, dass sich die UDI Daten in die Bereiche BASIC UDI-DI und UDI-DI aufteilen. Die BASIC UDI-DI dient dazu alle gemeinsamen Eigenschaften einer Produktgruppe abzubilden. Die UDI-DI enthält nur die produktspezifischen Informationen. Zu einem BASIC UDI-DI kann es mehrere UDI-DIs geben. Umgekehrt ist eine UDI-DI genau einem BASIC UDI-DI zugeordnet (siehe Abbildung unten).
Eigenschaften einer Produktgruppe abzubilden. Die UDI-DI enthält nur die produktspezifischen Informationen. Zu einem BASIC UDI-DI kann es mehrere UDI-DIs geben. Umgekehrt ist eine UDI-DI genau einem BASIC UDI-DI zugeordnet (siehe Abbildung unten).
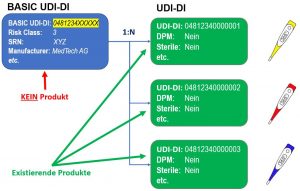
EUDAMED BASIC UDI-DI und UDI-DI
Als Wirtschaftsakteur ist man selbst dafür verantwortlich alle UDI Attribute im eigenen Unternehmen zu verwalten und die Daten zur EUDAMED zu übertragen. Lesen hier wie wir Sie dabei unterstützen können.


Datenfelder der EUDAMED – EUDAMED Excel Template
Weitere Informationen über das von uns entwickelte Excel Template für die UDI EUDAMED finden Sie hier.
Öffentlicher Zugang. Die Öffentlichkeit kann die SS(C)P (Zusammenfassung der Sicherheit [und klinischen] Leistung) einsehen, die einem registrierten Produkt beigefügt ist, registrierte Basis-UDI-DI- und UDI-DI-Daten herunterladen, Informationen über die registrierte Basis-UDI-DI, UDI-DI und das Produkt einsehen.
3. Modul CRF – Certificate 
Jedes Medizinprodukt welches in Europa vertrieben wird, benötigt zukünftig gemäss der MDR 2017/745 und IVDR 2017/746 ein gültiges Zulassungszertifikat. Dieses Zertifikat bescheinigt, dass das Produkt alle regulatorischen Anforderungen eines Medizinproduktes erfüllt. Durch die Zusammenfassung einer Produktgruppe in eine BASIC UDI-DI benötigt man lediglich ein Zertifikat für eine Produktgruppe anstatt für jeden Artikel innerhalb einer Produktgruppe. Diese Zertifikate werden künftig in dem Modul CRF verwaltet.
Öffentlicher Zugang zur Registrierung von Konformitätsbescheinigungen, deren Umfang und Gültigkeitsdauer.
4. Modul CIPS – Clinical Investigation

Nach der neuen Medizinprodukteverordnung (MDR) sind die Hersteller von Medizinprodukten verpflichtet, für alle ihre Produkte – unabhängig von der Risikoklasse – eine klinische Bewertung durchzuführen, die auch eine klinische Nachbeobachtung nach dem Inverkehrbringen (sog. Post Market Clinical Follow-up, PMCF) umfasst. Die klinische Bewertung ist eine wesentliche Herstelleraufgabe und fester Bestandteil des Qualitätsmanagementsystems eines Herstellers (Artikel 10, Abs. 3 und 9f MDR). Per Definition handelt es sich bei der klinischen Bewertung um einen systematischen und geplanten Prozess zur kontinuierlichen Generierung, Sammlung, Analyse und Bewertung der klinischen Daten zu einem Produkt. Durch die klinische Bewertung überprüft der Hersteller die Sicherheit und Leistung seines Produkts, einschließlich des klinischen Nutzens.
Teilweise öffentlich zugänglich. Die Registrierung von klinischen Prüfungen, die Berichterstattung über klinische Prüfergebnisse und die Veröffentlichung sind öffentlich zugänglich. Antragsdokumente für klinische Prüfungen werden potenziell öffentlich zugänglich sein.
5. Modul VGL – Vigilance

Jedes schwerwiegende Vorkommnis im Zusammenhang mit Medizinprodukten muss dokumentiert und bereitgestellt werden. In der technischen Dokumentation müssen Anzahl der Vorkommnisse und Gegenstand gemeldet werden. Sichherheit-Korrekturmassnahmen zu dem auf dem Unionsmarkt bereitgestellten Produkten müssen ebenso gemeldet werden.
Teilweiser öffentlicher Zugang für Hersteller Vorfallberichte und die Feldsicherheitshinweise für Vigilance Aktivitäten.
6. Modul MSU – Market Surveillance

Im Modul MSU werden die Ergebnisse der Marktüberwachungen zurückgemeldet. Dabei sind die zuständigen Behörden diejenigen, die die Marktüberwachungen vornehmen und die Berichte veröffentlichen. Die zuständige Behörde, die die Kontrolle durchgeführt hat, teilt dem Wirtschaftsakteur, der Gegenstand der Kontrolle war, den Inhalt des Berichts gemäss Absatz 6 des vorliegenden Artikels mit. Bevor die zuständige Behörde den Bericht annimmt, gibt sie diesem Wirtschaftsakteur Gelegenheit zur Stellungnahme. Dieser abschließende Kontrollbericht wird in dem in Artikel 100 vorgesehenen elektronischen System (Modul MSU) erfasst.
Zusammenfassung öffentlich zugänglich. Die Öffentlichkeit kann eine Zusammenfassung der Ergebnisse der Überprüfungen und Bewertungen der Marktüberwachungstätigkeiten einer Marktüberwachung einsehen.