By Ugur Müldür
In News

Time is running out for the medical device industry to comply with the requirements of the Medical Device Regulation (MDR). To properly implement such complex regulations, it’s essential to have a clear understanding of the various terms involved. In the UDI regulations alone, there are many key terms that need to be known. That’s why we’ve compiled over 20 important terms related to MDR/UDI to help ease your transition.
- Notified Body
An organization designated by an EU member state to carry out the conformity assessment of medical devices. The Notified Body evaluates whether a medical device meets the requirements of EU regulations before it is placed on the market. - Competent Authority
In each EU member state, there is a national authority responsible for monitoring and enforcing medical device regulations. They ensure that products comply with EU regulations and oversee the Notified Bodies. - Authorized Representative (AR)
An agent appointed by a manufacturer not based in the EU to act as a contact for fulfilling EU regulations. The AR is established in the EU and represents the manufacturer to authorities. - Manufacturer (MF)
The natural or legal person who develops, produces, and markets a medical device under their own name. The manufacturer is responsible for ensuring the product meets regulatory requirements. - Supplier
A company or person providing materials or components used in the production of a medical device. The supplier contributes to regulatory compliance by providing safe and compliant materials. - OEM Manufacturer (Original Equipment Manufacturer)
A manufacturer that produces products or components on behalf of other companies, who then sell them under their own name. In the medical technology field, the OEM manufacturer may also be responsible for regulatory compliance. - Importer (IM)
A person or organization that imports a medical device from a third country into the EU. The importer is responsible for ensuring the product complies with EU regulations before it is placed on the market. - System and Procedure Packer (SPP)
Companies or individuals responsible for packaging and labeling medical devices in accordance with requirements, including combining several products into a system. They must ensure that labeling is correct and packaging is safe. - Health Authority
National or international government agencies responsible for regulating and overseeing medical and health products. Examples include:- USA: Food and Drug Administration (FDA)
- EU: European Commission (EC)
- Canada: Therapeutic Goods Administration (TGA)
- China: National Medical Products Administration (NMPA)
- Economic Operator
A collective term for various actors in the supply chain of medical devices who play a regulatory role. This includes manufacturers, authorized representatives, importers, and distributors. Each economic operator has specific obligations under the EU Medical Device Regulation (MDR) or In Vitro Diagnostic Regulation (IVDR). - Distributor
A person or organization that distributes a medical device without altering it. The distributor must ensure that the product is properly labeled, has the necessary declarations of conformity, and meets regulatory requirements. - Declaration of Conformity
A formal declaration by the manufacturer that a medical device meets the requirements of relevant EU regulations (MDR/IVDR). This declaration is a prerequisite for CE marking of the product. - CE Marking
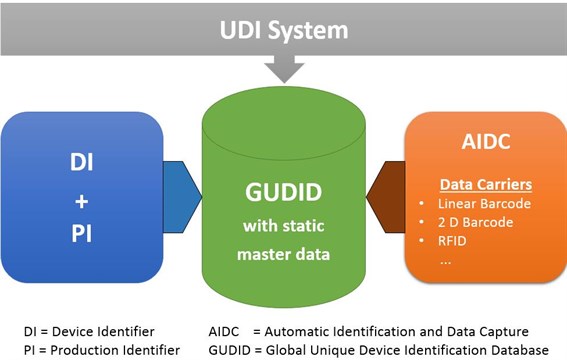
A marking that indicates a medical device complies with EU-wide regulations and can be legally placed on the European market. It is a sign of product conformity with EU health, safety, and environmental standards. - Unique Device Identification (UDI)
A globally standardized system for identifying medical devices. Each product is assigned a unique identifier, which is stored on the packaging and in the EUDAMED database. The UDI consists of a device identifier and a production identifier. - EUDAMED (European Database on Medical Devices)
A central European database that collects and shares information on medical devices, economic operators, clinical investigations, and vigilance (post-market surveillance). It is part of the MDR and IVDR frameworks and aims to enhance transparency and traceability of medical devices. - Vigilance (Post-Market Surveillance)
The process of monitoring medical devices after they have been placed on the market. Manufacturers and other economic operators are required to collect and evaluate data on the safety and performance of products to identify and mitigate potential risks. - Clinical Evaluation
A systematic process in which data on the safety and clinical performance of a medical device is collected and analyzed. This evaluation is required to confirm compliance with the regulatory requirements of the MDR. - Technical Documentation
A comprehensive collection of documents that the manufacturer must create to demonstrate the conformity of a medical device with relevant regulations. It includes information such as product design, risk management files, clinical evaluations, and instructions for use. - Risk Management
An ongoing process aimed at identifying, assessing, and minimizing potential risks associated with a medical device. This is a key part of the conformity assessment and is governed by international standards such as ISO 14971. - Certification Body
An organization that verifies and certifies compliance with international standards for quality management systems, such as ISO 13485. These certifications are often a prerequisite for market approval of medical devices. - Clinical Investigation
A study in which a medical device is tested on patients to evaluate its safety and effectiveness. Clinical trials are often required to gather data for the clinical evaluation and approval of the product.







Related Posts